

Using structural motif templates to identify proteins with DNA binding function
- PMID: 12771208
- PMCID: PMC156721
- DOI: 10.1093/nar/gkg386
Using structural motif templates to identify proteins with DNA binding function
Abstract
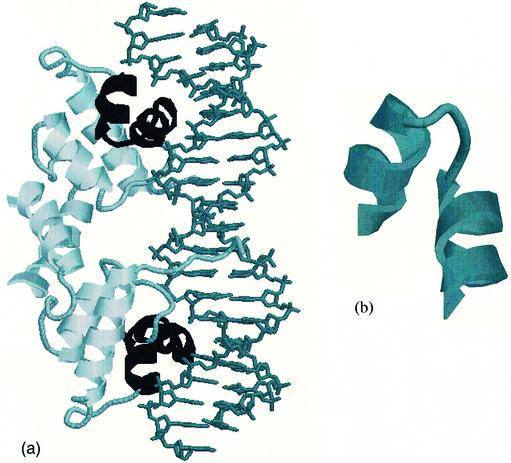
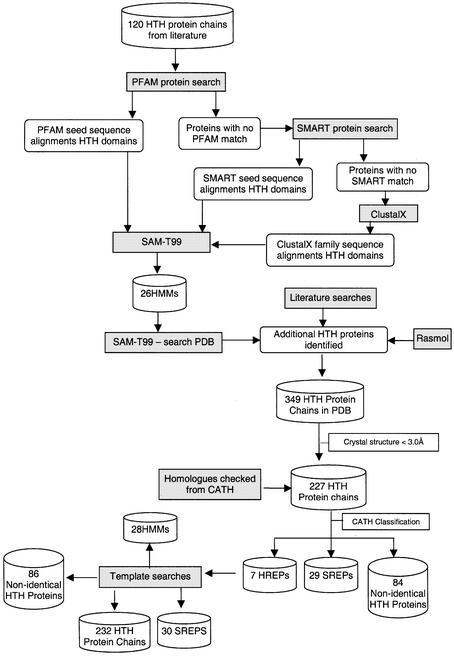
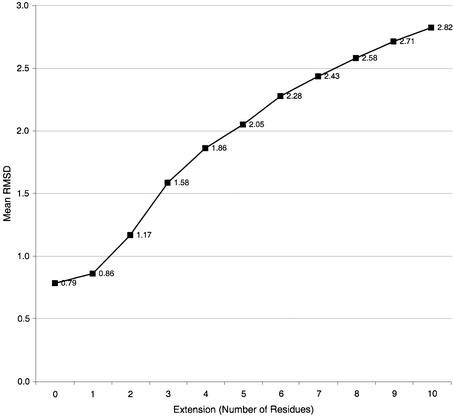
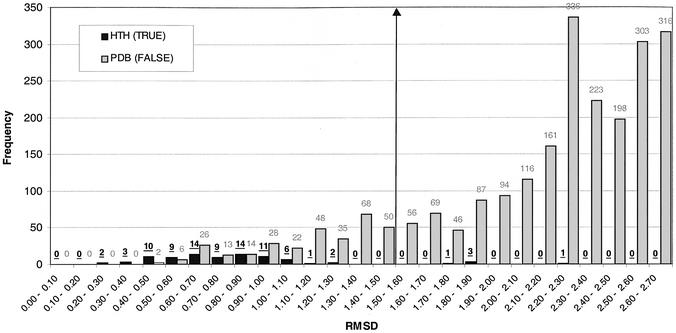
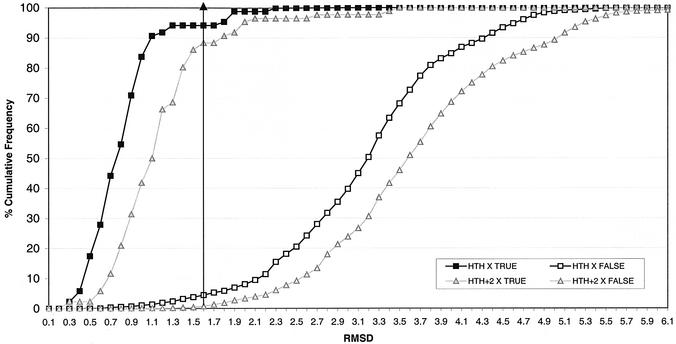
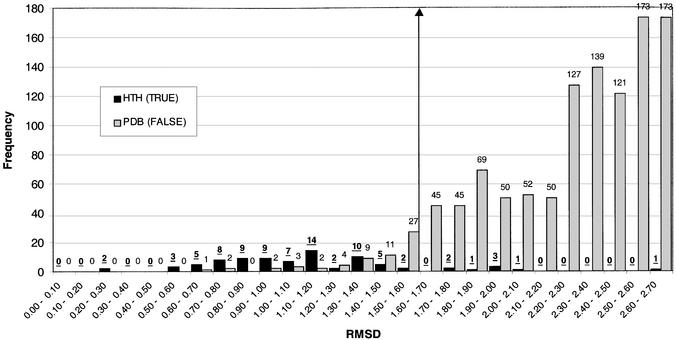
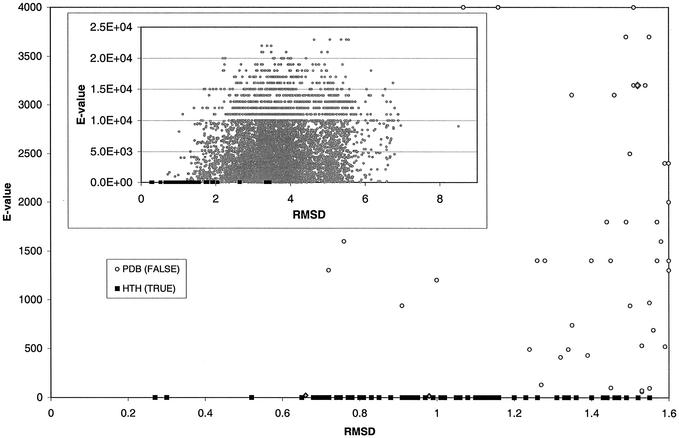
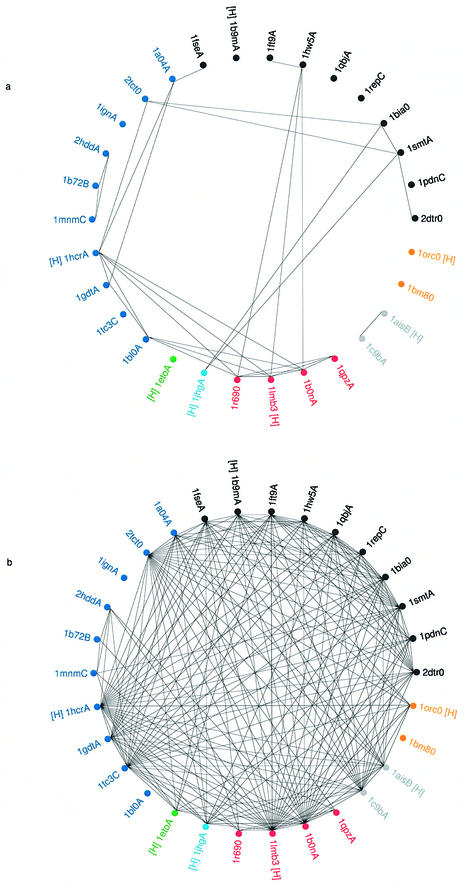
This work describes a method for predicting DNA binding function from structure using 3-dimensional templates. Proteins that bind DNA using small contiguous helix-turn-helix (HTH) motifs comprise a significant number of all DNA-binding proteins. A structural template library of seven HTH motifs has been created from non-homologous DNA-binding proteins in the Protein Data Bank. The templates were used to scan complete protein structures using an algorithm that calculated the root mean squared deviation (rmsd) for the optimal superposition of each template on each structure, based on C(alpha) backbone coordinates. Distributions of rmsd values for known HTH-containing proteins (true hits) and non-HTH proteins (false hits) were calculated. A threshold value of 1.6 A rmsd was selected that gave a true hit rate of 88.4% and a false positive rate of 0.7%. The false positive rate was further reduced to 0.5% by introducing an accessible surface area threshold value of 990 A2 per HTH motif. The template library and the validated thresholds were used to make predictions for target proteins from a structural genomics project.
Figures
References
-
- Brennan R.G. and Matthews,B.W. (1989) The helix-turn-helix DNA-binding motif. J. Biol. Chem., 264, 1903–1906. - PubMed
-
- Beamer L.J. (1992) Refined 1.8 angstrom crystal-structure of the lambda-repressor operator complex. J. Mol. Biol., 227, 20. - PubMed
-
- Luscombe N.M. and Thornton,J.M. (2002) Protein-DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J. Mol. Biol., 320, 991–1009. - PubMed
-
- Orengo C.A., Michie,A.D., Jones,S., Jones,D.T., Swindells,M.B. and Thornton,J.M. (1997) CATH – a hierarchic classification of protein domain structures. Structure, 5, 1093–1108. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
