

Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter
- PMID: 12773551
- PMCID: PMC156121
- DOI: 10.1128/MCB.23.12.4056-4065.2003
Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter
Abstract
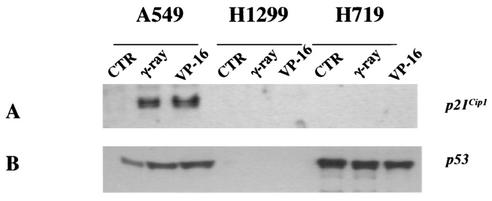
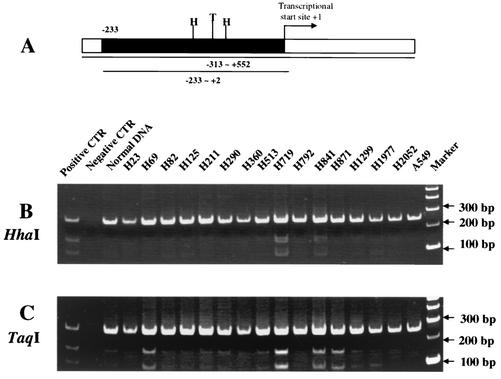
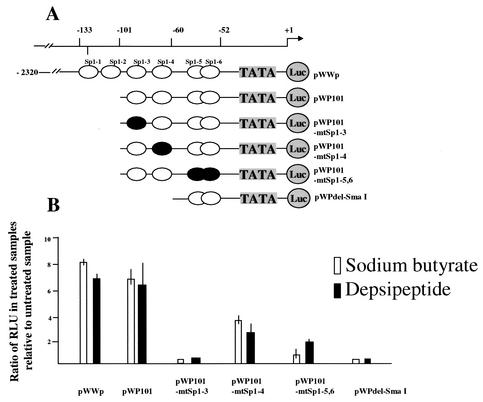
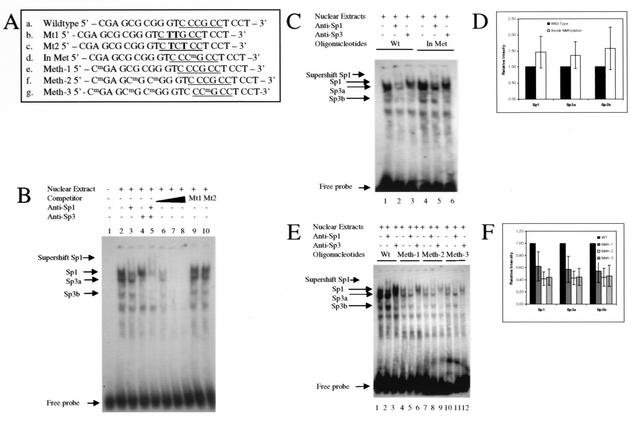
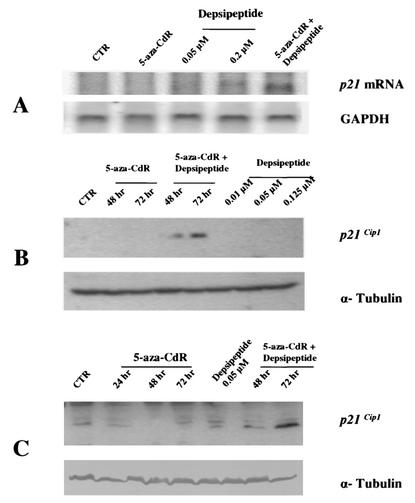
DNA methylation in the promoter of certain genes is associated with transcriptional silencing. Methylation affects gene expression directly by interfering with transcription factor binding and/or indirectly by recruiting histone deacetylases through methyl-DNA-binding proteins. In this study, we demonstrate that the human lung cancer cell line H719 lacks p53-dependent and -independent p21(Cip1) expression. p53 response to treatment with gamma irradiation or etoposide is lost due to a mutation at codon 242 of p53 (C-->W). Treatment with depsipeptide, an inhibitor of histone deacetylase, was unable to induce p53-independent p21(Cip1) expression because the promoter of p21(Cip1) in these cells is hypermethylated. By analyzing luciferase activity of transfected p21(Cip1) promoter vectors, we demonstrate that depsipeptide functions on Sp1-binding sites to induce p21(Cip1) expression. We hypothesize that hypermethylation may interfere with Sp1/Sp3 binding. By using an electrophoretic mobility shift assay, we show that, although methylation within the consensus Sp1-binding site did not reduce Sp1/Sp3 binding, methylation outside of the consensus Sp1 element induced a significant decrease in Sp1/Sp3 binding. Depsipeptide induced p21(Cip1) expression was reconstituted when cells were pretreated with 5-aza-2'-deoxycytidine. Our data suggest, for the first time, that hypermethylation around the consensus Sp1-binding sites may directly reduce Sp1/Sp3 binding, therefore leading to a reduced p21(Cip1) expression in response to depsipeptide treatment.
Figures


References
-
- Alland, L., R. Muhle, H. Hou, Jr., J. Potes, L. Chin, N. Schreiber-Agus, and R. A. DePinho. 1997. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature 387:49-55. - PubMed
-
- Baylin, S. B., M. Esteller, M. R. Rountree, K. E. Bachman, K. Schuebel, and J. G. Herman. 2001. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 10:687-692. - PubMed
-
- Berger, S. L. 2002. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12:142-148. - PubMed
-
- Bird, A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16:6-21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous