

Prophage genomics
- PMID: 12794192
- PMCID: PMC156470
- DOI: 10.1128/MMBR.67.2.238-276.2003
Prophage genomics
Erratum in
- Microbiol Mol Biol Rev. 2003 Sep;67(3):473
Abstract
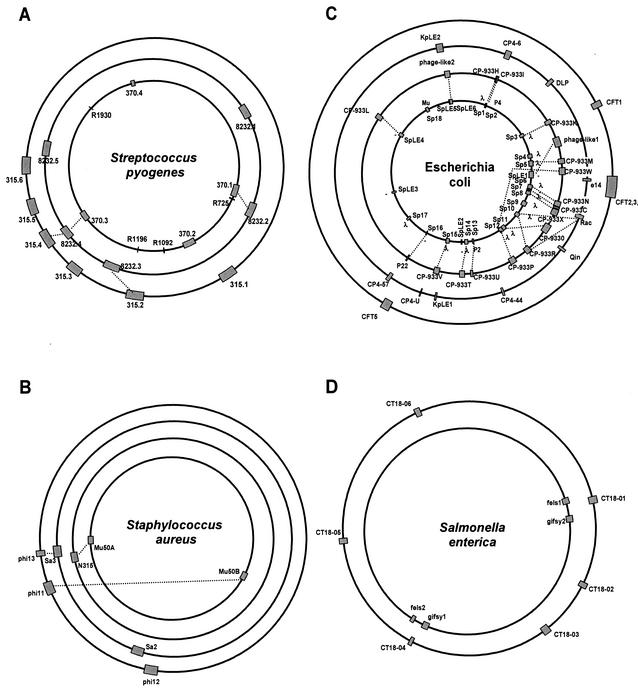
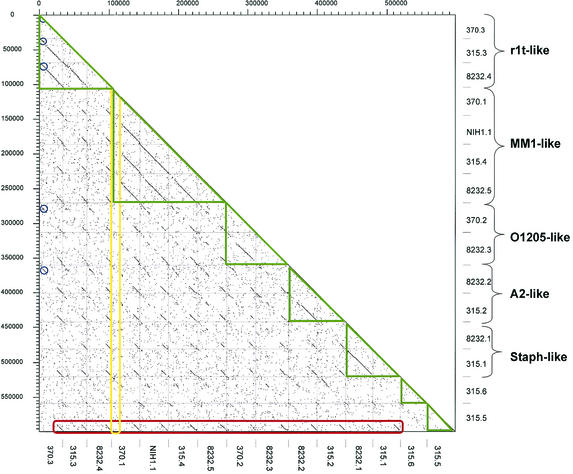
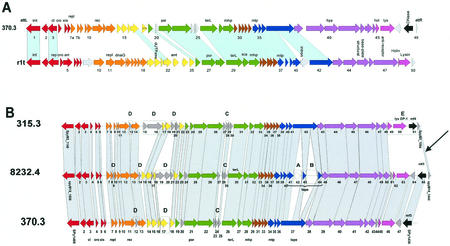
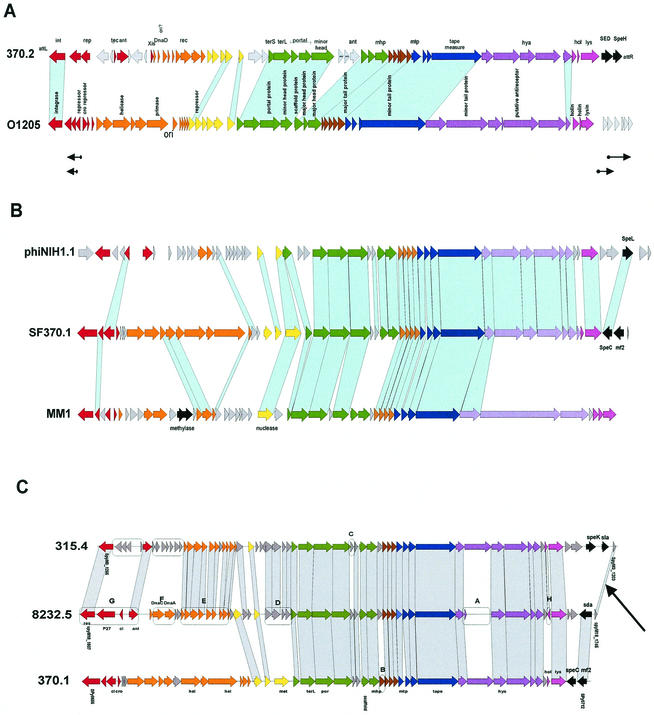
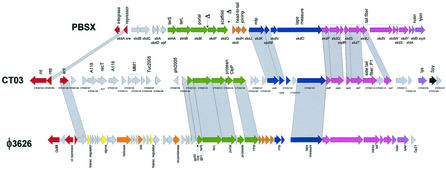
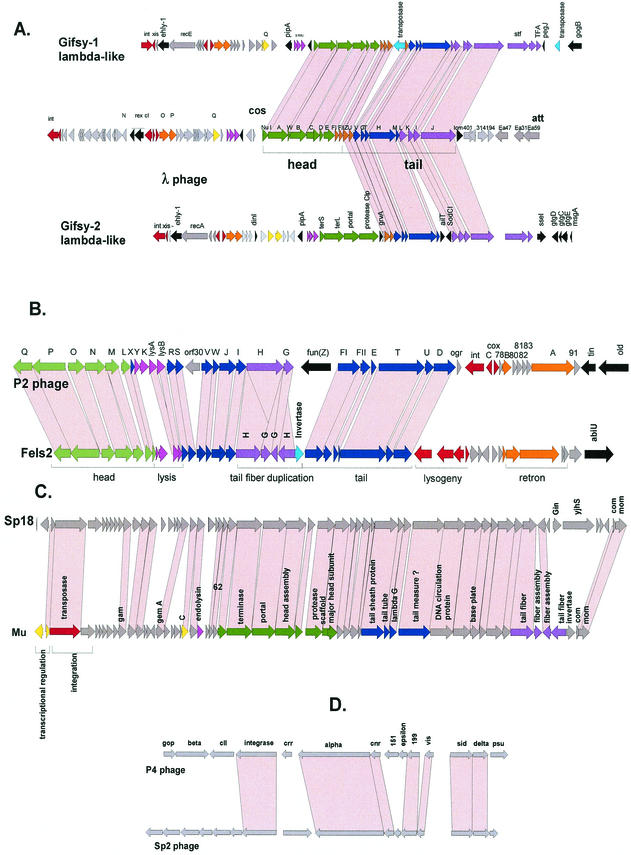
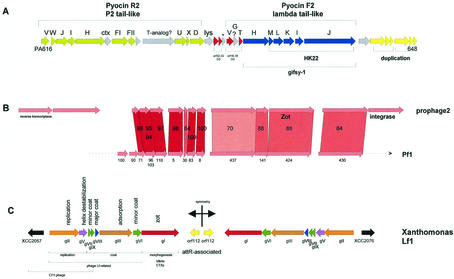
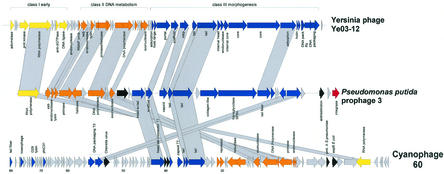
The majority of the bacterial genome sequences deposited in the National Center for Biotechnology Information database contain prophage sequences. Analysis of the prophages suggested that after being integrated into bacterial genomes, they undergo a complex decay process consisting of inactivating point mutations, genome rearrangements, modular exchanges, invasion by further mobile DNA elements, and massive DNA deletion. We review the technical difficulties in defining such altered prophage sequences in bacterial genomes and discuss theoretical frameworks for the phage-bacterium interaction at the genomic level. The published genome sequences from three groups of eubacteria (low- and high-G+C gram-positive bacteria and gamma-proteobacteria) were screened for prophage sequences. The prophages from Streptococcus pyogenes served as test case for theoretical predictions of the role of prophages in the evolution of pathogenic bacteria. The genomes from further human, animal, and plant pathogens, as well as commensal and free-living bacteria, were included in the analysis to see whether the same principles of prophage genomics apply for bacteria living in different ecological niches and coming from distinct phylogenetical affinities. The effect of selection pressure on the host bacterium is apparently an important force shaping the prophage genomes in low-G+C gram-positive bacteria and gamma-proteobacteria.
Figures
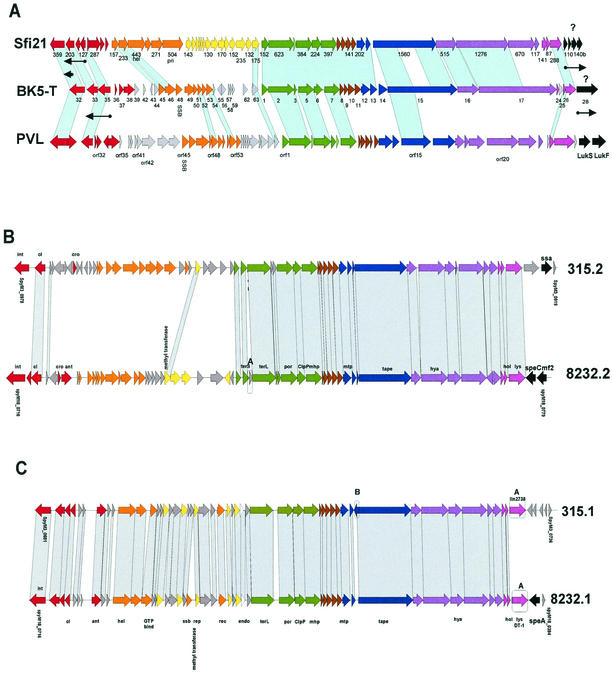
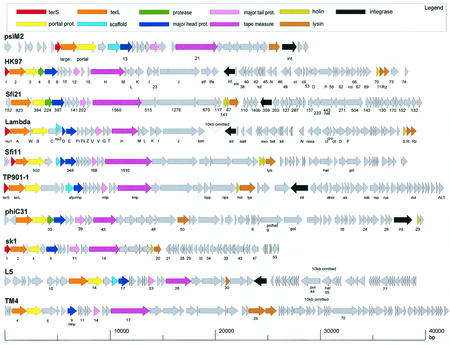
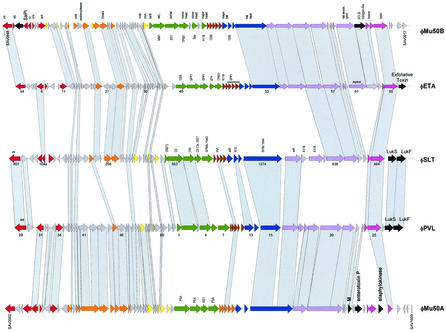
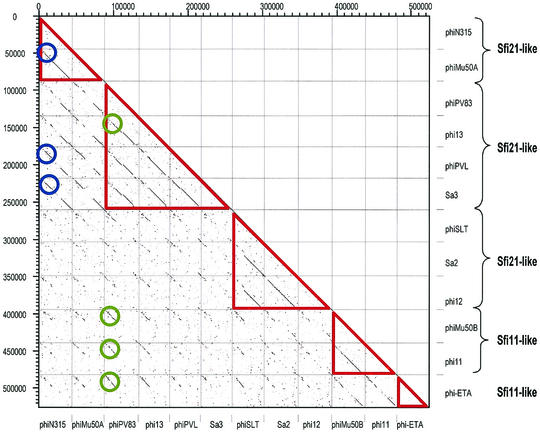
References
-
- Akman, L., A. Yamashita, H. Watanabe, K. Oshima, T. Shiba, M. Hattori, and S. Aksoy. 2002. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 32:402-407. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
