

Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency
- PMID: 12809642
- PMCID: PMC2813192
- DOI: 10.1016/s1096-7192(03)00073-8
Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency
Abstract
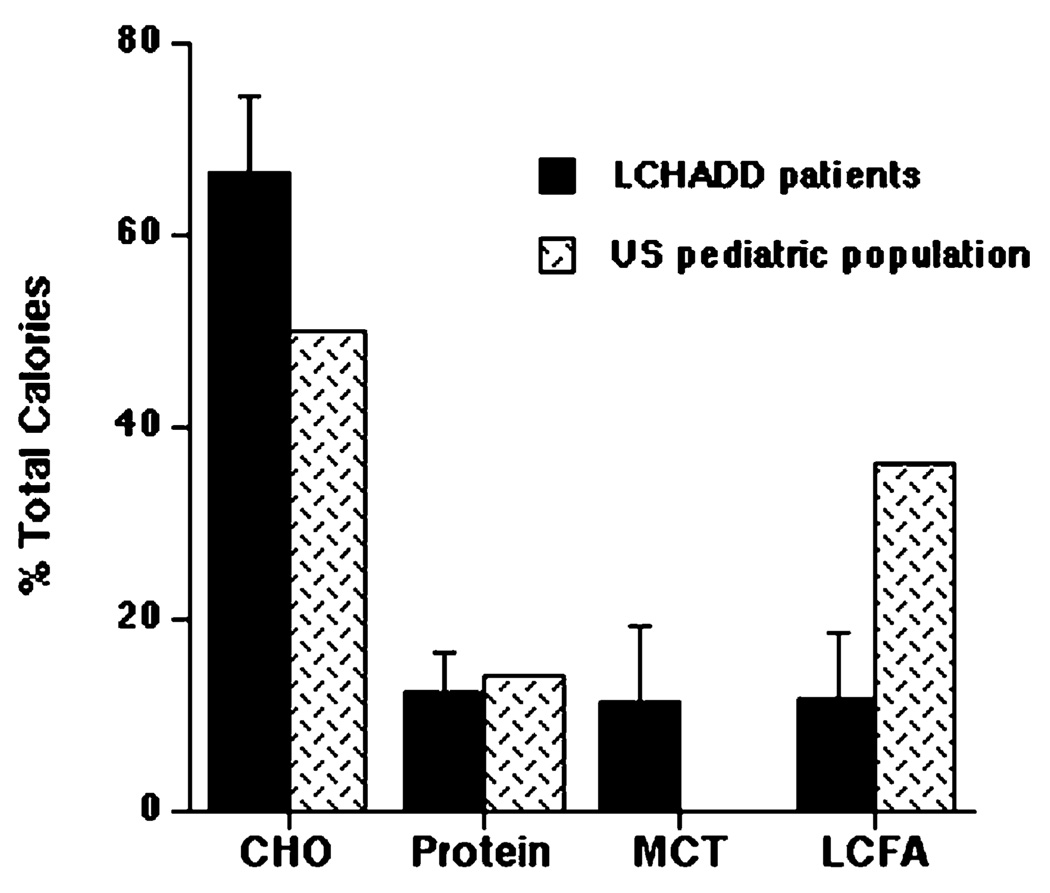
Current dietary therapy for long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency consists of fasting avoidance, and limiting long-chain fatty acid (LCFA) intake. This study reports the relationship of dietary intake and metabolic control as measured by plasma acylcarnitine and organic acid profiles in 10 children with LCHAD or TFP deficiency followed for 1 year. Subjects consumed an average of 11% of caloric intake as dietary LCFA, 11% as MCT, 12% as protein, and 66% as carbohydrate. Plasma levels of hydroxypalmitoleic acid, hydroxyoleic, and hydroxylinoleic carnitine esters positively correlated with total LCFA intake and negatively correlated with MCT intake suggesting that as dietary intake of LCFA decreases and MCT intake increases, there is a corresponding decrease in plasma hydroxyacylcarnitines. There was no correlation between plasma acylcarnitines and level of carnitine supplementation. Dietary intake of fat-soluble vitamins E and K was deficient. Dietary intake and plasma levels of essential fatty acids, linoleic and linolenic acid, were deficient. On this dietary regimen, the majority of subjects were healthy with no episodes of metabolic decompensation. Our data suggest that an LCHAD or TFP-deficient patient should adhere to a diet providing age-appropriate protein and limited LCFA intake (10% of total energy) while providing 10-20% of energy as MCT and a daily multi-vitamin and mineral (MVM) supplement that includes all of the fat-soluble vitamins. The diet should be supplemented with vegetable oils as part of the 10% total LCFA intake to provide essential fatty acids.
Figures




References
-
- Costa CG, Dorland L, Holwerda U, de Almeida IT, Poll-The BT, Jakobs C, Duran M. Simultaneous analysis of plasma free fatty acids and their 3-hydroxy analogs in fatty acid β-oxidation disorders. Clin. Chem. 1998;44:463–471. - PubMed
-
- Van Hove JL, Kahler SG, Feezor MD, Ramakrishna JP, Hart P, Treem WR, Shen JJ, Matern D, Millington DS. Acylcarnitines in plasma and blood spots of patients with long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. J. Inherit. Metab. Dis. 2000;23:571–582. - PubMed
-
- Saudubray JM, Martin D, de Lonlay P, Touati G, Poggi-Travert F, Bonnet D, Jouvet P, Boutron M, Slama A, Vianey-Saban C, Bonnefont JP, Rabier D, Kamoun P, Brivet M. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J. Inherit. Metab. Dis. 1999;22:488–502. - PubMed
-
- Ibdah JA, Bennett MJ, Rinaldo P, Zhao Y, Gibson B, Sims HF, Strauss AW. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N. Engl. J. Med. 1999;340:1723–1731. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
