

CDS annotation in full-length cDNA sequence
- PMID: 12819146
- PMCID: PMC403693
- DOI: 10.1101/gr.1060303
CDS annotation in full-length cDNA sequence
Abstract
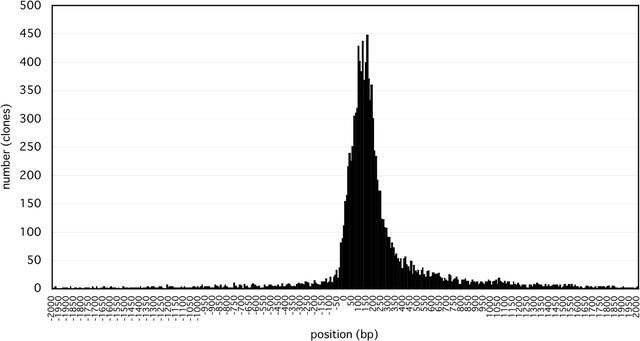
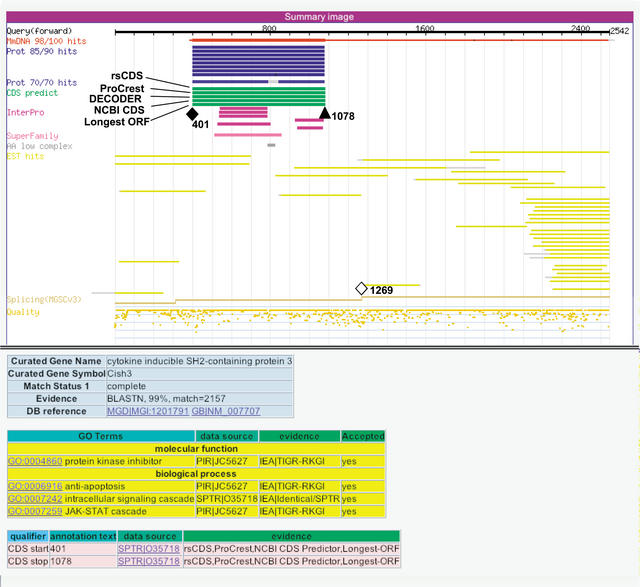
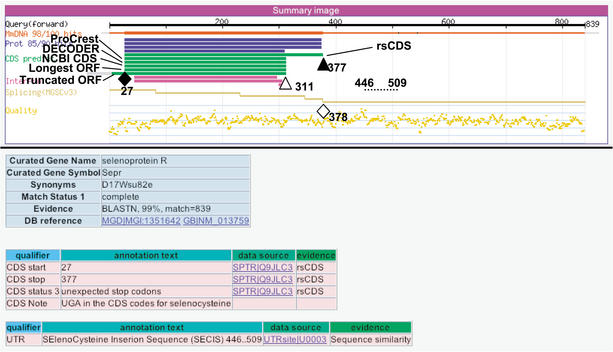
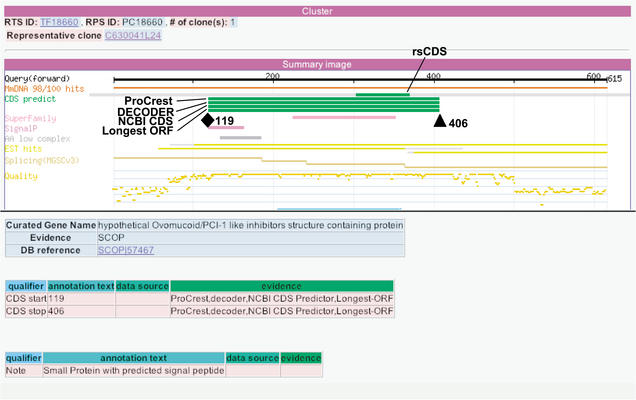
The identification of coding sequences (CDS) is an important step in the functional annotation of genes. CDS prediction for mammalian genes from genomic sequence is complicated by the vast abundance of intergenic sequence in the genome, and provides little information about how different parts of potential CDS regions are expressed. In contrast, mammalian gene CDS prediction from cDNA sequence offers obvious advantages, yet encounters a different set of complexities when performed on high-throughput cDNA (HTC) sequences, such as the set of 60,770 cDNAs isolated from full-length enriched libraries of the FANTOM2 project. We developed a CDS annotation strategy that uses a variety of different CDS prediction programs to annotate the CDS regions of FANTOM2 cDNAs. These include rsCDS, which uses sequence similarity to known proteins; ProCrest; Longest-ORF and Truncated-ORF, which are ab initio based predictors; and finally, DECODER and NCBI CDS predictor, which use a combination of both principles. Aided by graphical displays of these CDS prediction results in the context of other sequence similarity results for each cDNA, FANTOM2 CDS inspection by curators and follow-up quality control procedures resulted in high quality CDS predictions for a total of 14,345 FANTOM2 clones.
Figures



References
-
- Berry, M.J., Banu, L., Chen, Y.Y., Mandel, S.J., Kieffer, J.D., Harney, J.W., and Larsen, P.R. 1991. Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3′ untranslated region. Nature 353: 273-276. - PubMed
-
- The FANTOM Consortium and the RIKEN Genome Exploration Research Group Phase I & II Team. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563-573. - PubMed
-
- Fukunishi, Y. and Hayashizaki, Y. 2001. Amino acid translation program for full-length cDNA sequences with frameshift errors. Physiol. Genom. 5: 81-87. - PubMed
-
- Hentze, M.W. and Kulozik, A.E. 1999. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 96: 307-310. - PubMed
WEB SITE REFERENCES
-
- http://fantom2.gsc.riken.go.jp/; FANTOM2.
-
- http://fantom2.gsc.riken.go.jp/db/; FANTOM2 cDNA annotation database.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases