

STING Millennium: A web-based suite of programs for comprehensive and simultaneous analysis of protein structure and sequence
- PMID: 12824333
- PMCID: PMC168984
- DOI: 10.1093/nar/gkg578
STING Millennium: A web-based suite of programs for comprehensive and simultaneous analysis of protein structure and sequence
Abstract
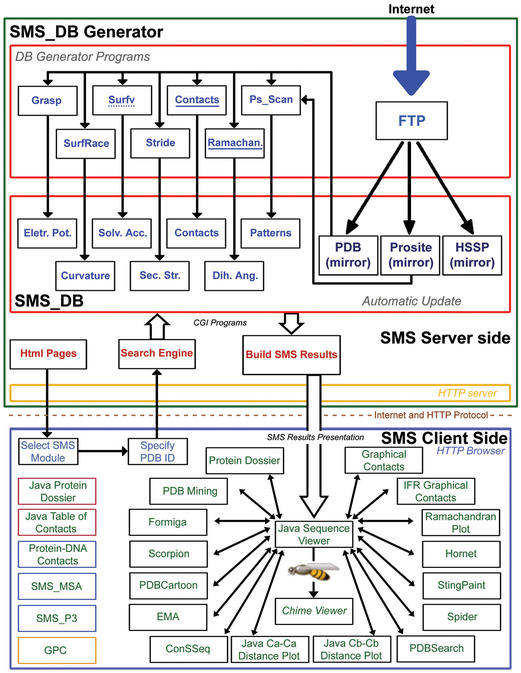
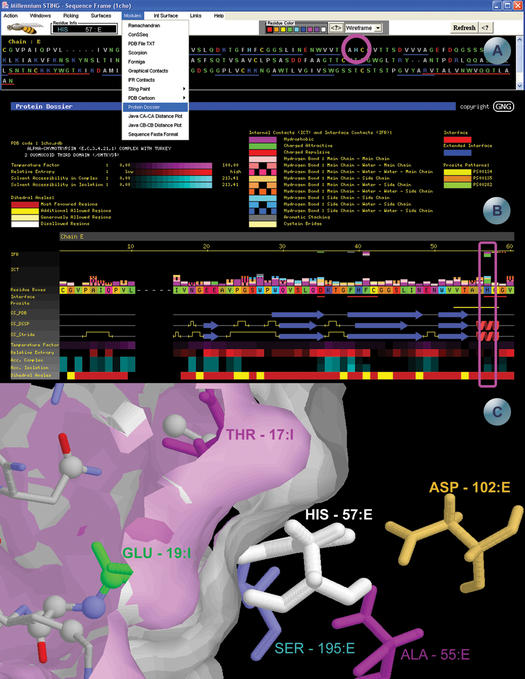
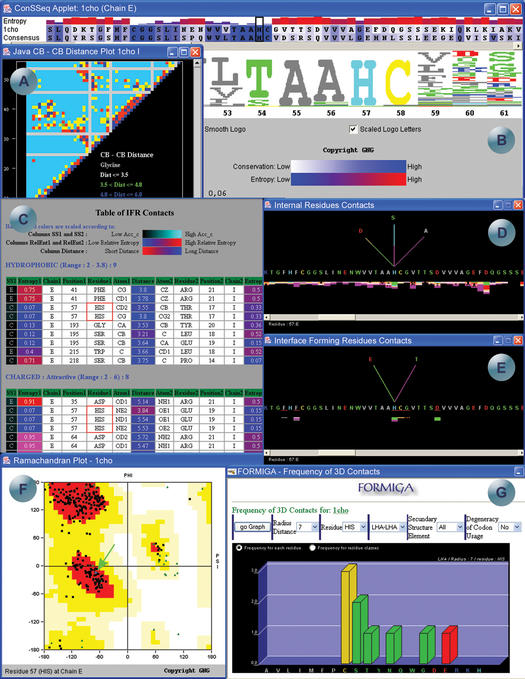
STING Millennium Suite (SMS) is a new web-based suite of programs and databases providing visualization and a complex analysis of molecular sequence and structure for the data deposited at the Protein Data Bank (PDB). SMS operates with a collection of both publicly available data (PDB, HSSP, Prosite) and its own data (contacts, interface contacts, surface accessibility). Biologists find SMS useful because it provides a variety of algorithms and validated data, wrapped-up in a user friendly web interface. Using SMS it is now possible to analyze sequence to structure relationships, the quality of the structure, nature and volume of atomic contacts of intra and inter chain type, relative conservation of amino acids at the specific sequence position based on multiple sequence alignment, indications of folding essential residue (FER) based on the relationship of the residue conservation to the intra-chain contacts and Calpha-Calpha and Cbeta-Cbeta distance geometry. Specific emphasis in SMS is given to interface forming residues (IFR)-amino acids that define the interactive portion of the protein surfaces. SMS may simultaneously display and analyze previously superimposed structures. PDB updates trigger SMS updates in a synchronized fashion. SMS is freely accessible for public data at http://www.cbi.cnptia.embrapa.br, http://mirrors.rcsb.org/SMS and http://trantor.bioc.columbia.edu/SMS.
Figures
References
-
- Laskovski R.A., Hutchinson,E.G., Michie,A.D., Wallace,A.C., Jones,M.L. and Thornton,J.M. (1997) PDBsum: a web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci., 22, 488–490. - PubMed
-
- Hogue C.W. (1997) Cn3D: a new generation of three-dimensional molecular structure viewer. Trends Biochem. Sci., 22, 314–316. - PubMed
-
- Neshich G., Togawa,R.C. and Honig,B. (1998) Sequence To and withIN graphics PDB Viewer (STING-PDB viewer). PDB Quart. News., July (electronic edition), 6.
-
- Sander C. and Schneider,R. (1991) Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins, 9, 56–68. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
