

doi: 10.1093/nar/gkg500.
Multiple sequence alignment with the Clustal series of programs
Affiliations
- PMID: 12824352
- PMCID: PMC168907
- DOI: 10.1093/nar/gkg500
Item in Clipboard
Multiple sequence alignment with the Clustal series of programs
Nucleic Acids Res.
.
Abstract
The Clustal series of programs are widely used in molecular biology for the multiple alignment of both nucleic acid and protein sequences and for preparing phylogenetic trees. The popularity of the programs depends on a number of factors, including not only the accuracy of the results, but also the robustness, portability and user-friendliness of the programs. New features include NEXUS and FASTA format output, printing range numbers and faster tree calculation. Although, Clustal was originally developed to run on a local computer, numerous Web servers have been set up, notably at the EBI (European Bioinformatics Institute) (http://www.ebi.ac.uk/clustalw/).
Figures
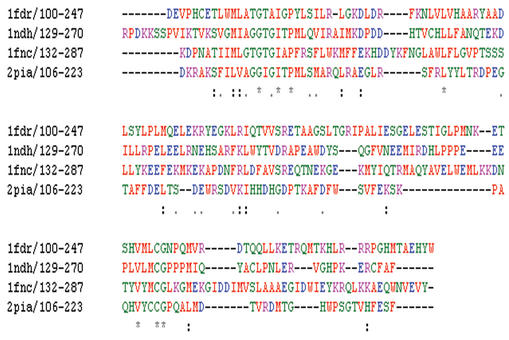
A multiple alignment of four oxidoreductase NAD binding domain protein sequences. Residues are coloured according to the following criteria: AVFPMILW are shown in red, DE are blue, RHK are magenta, STYHCNGQ are green and all other residues are grey. The residue range for each sequence is shown after the sequence name.
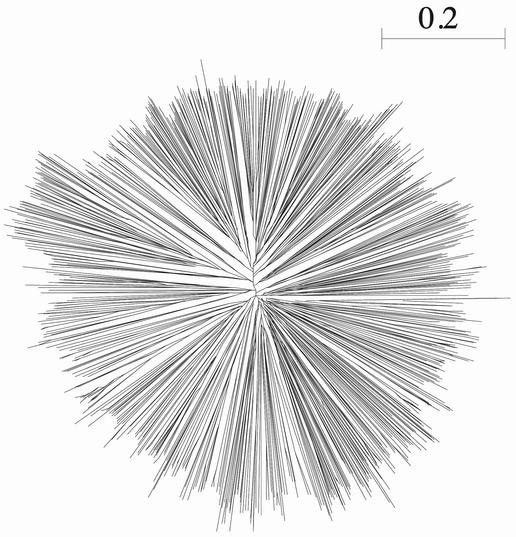
A tree calculated from an alignment of more than 1100 ring finger domains, using ClustalW 1.83. The full tree calculation, including the distance matrix calculation, took 22 s on a 1 GHz Pentium III. The output tree was displayed with Unrooted (18).
References
-
- Higgins D.G. and Sharp,P.M. (1988) CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene, 73, 237–244. - PubMed
-
- Myers E.W. and Miller,W. (1988) Optimal alignments in linear space. Comput. Applic. Biosci., 4, 11–17. - PubMed
-
- Feng D.F. and Doolittle,R.F. (1987) Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol., 25, 351–360. - PubMed
-
- Taylor W.R. (1988) A flexible method to align large numbers of biological sequences. J. Mol. Evol., 28, 161–169. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
