

MultiPipMaker and supporting tools: Alignments and analysis of multiple genomic DNA sequences
- PMID: 12824357
- PMCID: PMC168985
- DOI: 10.1093/nar/gkg579
MultiPipMaker and supporting tools: Alignments and analysis of multiple genomic DNA sequences
Abstract
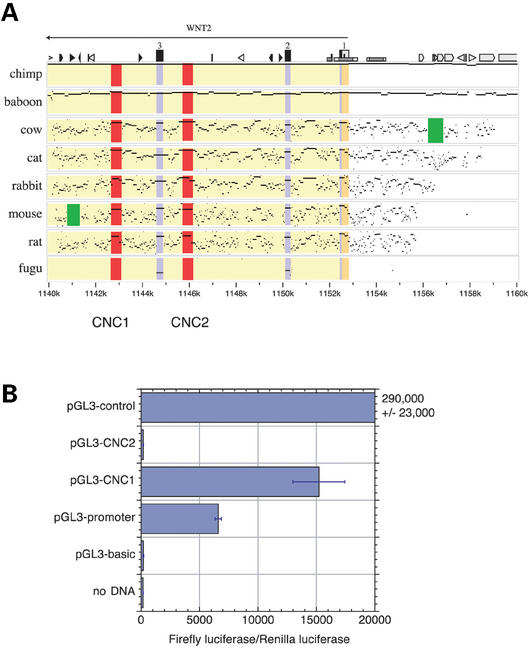
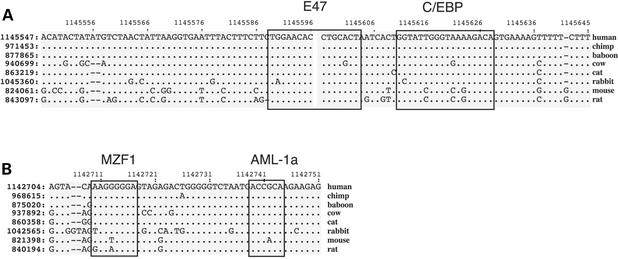
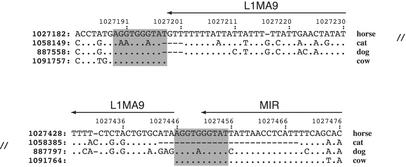
Analysis of multiple sequence alignments can generate important, testable hypotheses about the phylogenetic history and cellular function of genomic sequences. We describe the MultiPipMaker server, which aligns multiple, long genomic DNA sequences quickly and with good sensitivity (available at http://bio.cse.psu.edu/ since May 2001). Alignments are computed between a contiguous reference sequence and one or more secondary sequences, which can be finished or draft sequence. The outputs include a stacked set of percent identity plots, called a MultiPip, comparing the reference sequence with subsequent sequences, and a nucleotide-level multiple alignment. New tools are provided to search MultiPipMaker output for conserved matches to a user-specified pattern and for conserved matches to position weight matrices that describe transcription factor binding sites (singly and in clusters). We illustrate the use of MultiPipMaker to identify candidate regulatory regions in WNT2 and then demonstrate by transfection assays that they are functional. Analysis of the alignments also confirms the phylogenetic inference that horses are more closely related to cats than to cows.
Figures

References
-
- Kimura M. (1977) Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature, 267, 275–276. - PubMed
-
- Li W.H., Gojobori,T. and Nei,M. (1981) Pseudogenes as a paradigm of neutral evolution. Nature, 292, 237–239. - PubMed
-
- Pennacchio L.A. and Rubin,E.M. (2001) Genomic strategies to identify mammalian regulatory sequences. Nature Rev. Genet., 2, 100–109. - PubMed
-
- Li W., Ellsworth,D., Krushkal,J., Chang,B. and Hewett-Emmett,D. (1996) Rates of nucleotide substitution in primates and rodents and the generation-time effect hypothesis. Mol. Phylogenet. Evol., 5, 182–187. - PubMed
-
- Wolfe K.H., Sharp,P.M. and Li,W.H. (1989) Mutation rates differ among regions of the mammalian genome. Nature, 337, 283–285. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
