

Physiological modulation of inactivation in L-type Ca2+ channels: one switch
- PMID: 12824441
- PMCID: PMC1664755
- DOI: 10.1113/jphysiol.2003.047902
Physiological modulation of inactivation in L-type Ca2+ channels: one switch
Abstract
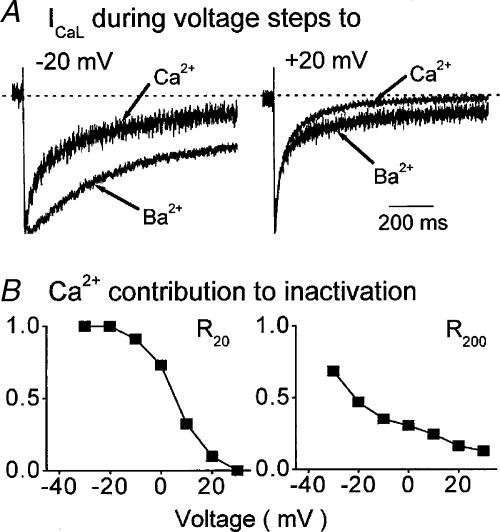
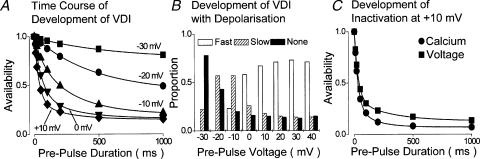
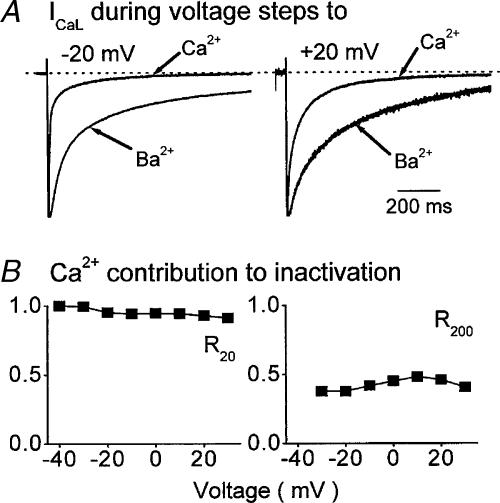
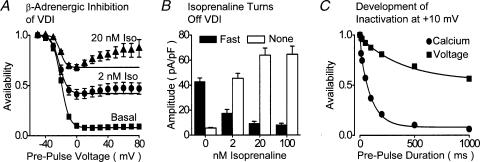
The relative contributions of voltage- and Ca(2+)-dependent mechanisms of inactivation to the decay of L-type Ca(2+) channel currents (I(CaL)) is an old story to which recent results have given an unexpected twist. In cardiac myocytes voltage-dependent inactivation (VDI) was thought to be slow and Ca(2+)-dependent inactivation (CDI) resulting from Ca(2+) influx and Ca(2+)-induced Ca(2+)-release (CICR) from the sarcoplasmic reticulum provided an automatic negative feedback mechanism to limit Ca(2+) entry and the contribution of I(CaL) to the cardiac action potential. Physiological modulation of I(CaL) by Beta-adrenergic and muscarinic agonists then involved essentially more or less of the same by enhancing or reducing Ca(2+) channel activity, Ca(2+) influx, sarcoplasmic reticulum load and thus CDI. Recent results on the other hand place VDI at the centre of the regulation of I(CaL). Under basal conditions it has been found that depolarization increases the probability that an ion channel will show rapid VDI. This is prevented by Beta-adrenergic stimulation. Evidence also suggests that a channel which shows rapid VDI inactivates before CDI can become effective. Therefore the contributions of VDI and CDI to the decay of I(CaL) are determined by the turning on, by depolarization, and the turning off, by phosphorylation, of the mechanism of rapid VDI. The physiological implications of these ideas are that under basal conditions the contribution of I(CaL) to the action potential will be determined largely by voltage and by Ca(2+) following Beta-adrenergic stimulation.
Figures
References
-
- Anderson ME. Ca2+-dependent regulation of cardiac L-type Ca2+ channels: is a unifying mechanism at hand. J Mol Cell Cardiol. 2001;33:639–650. - PubMed
-
- Antz C, Bauer T, Kalbacher H, Frank R, Covarrubias M, Kalbitzer HR, Ruppersberg JP, Baukrowitz T, Fakler B. Control of K+ channel gating by protein phosphorylation: structural switches of the inactivation gate. Nature Struc Biol. 1999;6:146–150. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous