

An evolutionarily structured universe of protein architecture
- PMID: 12840035
- PMCID: PMC403752
- DOI: 10.1101/gr.1161903
An evolutionarily structured universe of protein architecture
Abstract
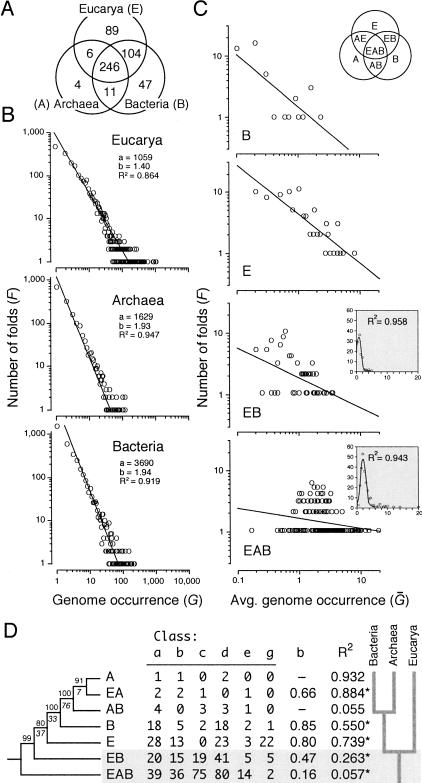
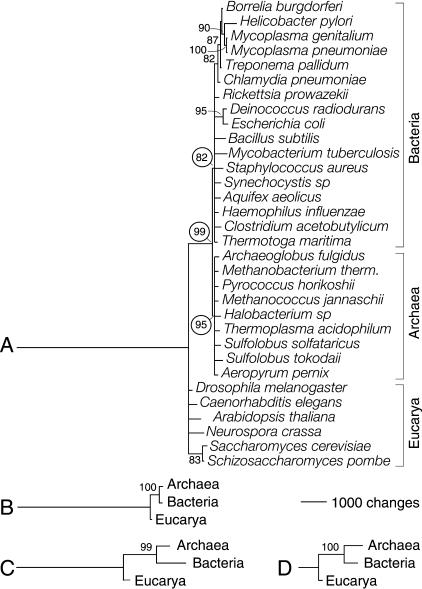
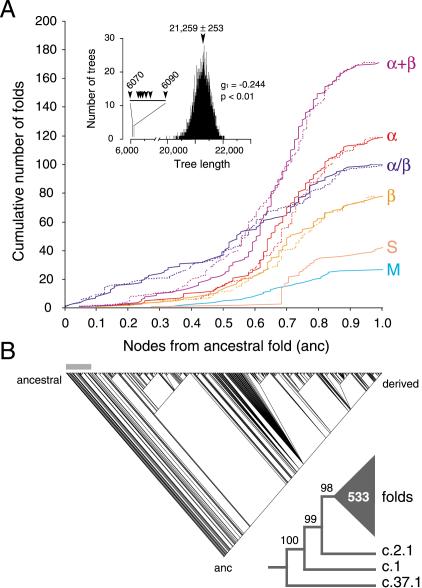
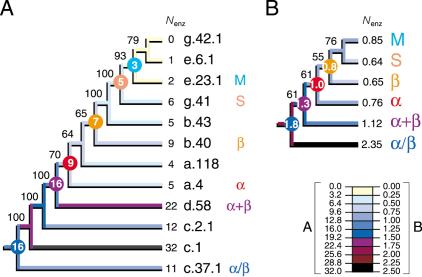
Protein structural diversity encompasses a finite set of architectural designs. Embedded in these topologies are evolutionary histories that we here uncover using cladistic principles and measurements of protein-fold usage and sharing. The reconstructed phylogenies are inherently rooted and depict histories of protein and proteome diversification. Proteome phylogenies showed two monophyletic sister-groups delimiting Bacteria and Archaea, and a topology rooted in Eucarya. This suggests three dramatic evolutionary events and a common ancestor with a eukaryotic-like, gene-rich, and relatively modern organization. Conversely, a general phylogeny of protein architectures showed that structural classes of globular proteins appeared early in evolution and in defined order, the alpha/beta class being the first. Although most ancestral folds shared a common architecture of barrels or interleaved beta-sheets and alpha-helices, many were clearly derived, such as polyhedral folds in the all-alpha class and beta-sandwiches, beta-propellers, and beta-prisms in all-beta proteins. We also describe transformation pathways of architectures that are prevalently used in nature. For example, beta-barrels with increased curl and stagger were favored evolutionary outcomes in the all-beta class. Interestingly, we found cases where structural change followed the alpha-to-beta tendency uncovered in the tree of architectures. Lastly, we traced the total number of enzymatic functions associated with folds in the trees and show that there is a general link between structure and enzymatic function.
Figures


References
-
- Ancel, L.W. and Fontana, W. 2000. Plasticity, evolvability, and modularity in RNA. J. Exp. Zool. Part B. Mol. Dev. Evol. 288: 242-283. - PubMed
-
- Apic, G., Gough, J., and Teichmann, S.A. 2001. An insight into domain combinations. Bioinformatics 17: S83-S89. - PubMed
-
- Aravind, L., Mazumder, R., Vasudevan, S., and Koonin, E.V. 2002a. Trends in protein evolution inferred from sequence and structure analysis. Curr. Opin. Struct. Biol. 12: 392-399. - PubMed
-
- Aravind, L., Anantharaman, V., and Koonin, E.V. 2002b. Monophily of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: Implications for protein evolution in the RNA world. Proteins 48: 1-14. - PubMed
-
- Bailey, J.A., Gu, Z., Clark, R.A., Reinert, K., Samonte, R.V., Schwartz, S., Adams, M.D., Myers, E.W., Li, P.W., and Eichler, E.E. 2002. Recent segmental duplications in the human genome. Science 297: 1003-1007. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources