

Abnormal passive chloride absorption in cystic fibrosis jejunum functionally opposes the classic chloride secretory defect
- PMID: 12840066
- PMCID: PMC162286
- DOI: 10.1172/JCI17667
Abnormal passive chloride absorption in cystic fibrosis jejunum functionally opposes the classic chloride secretory defect
Abstract
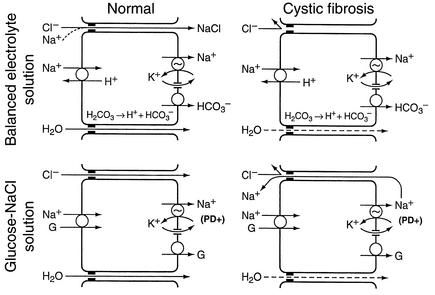
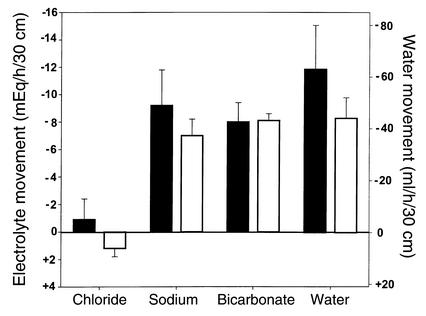
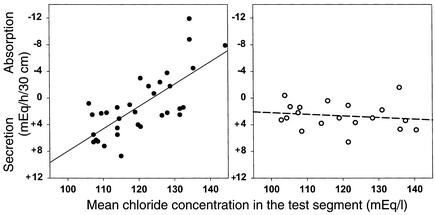
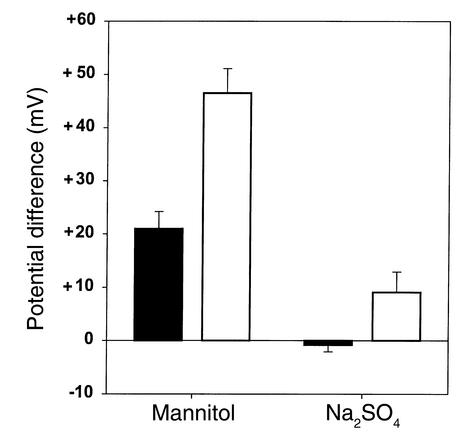
Due to genetic defects in apical membrane chloride channels, the cystic fibrosis (CF) intestine does not secrete chloride normally. Depressed chloride secretion leaves CF intestinal absorptive processes unopposed, which results in net fluid hyperabsorption, dehydration of intestinal contents, and a propensity to inspissated intestinal obstruction. This theory is based primarily on in vitro studies of jejunal mucosa. To determine if CF patients actually hyperabsorb fluid in vivo, we measured electrolyte and water absorption during steady-state perfusion of the jejunum. As expected, chloride secretion was abnormally low in CF, but surprisingly, there was no net hyperabsorption of sodium or water during perfusion of a balanced electrolyte solution. This suggested that fluid absorption processes are reduced in CF jejunum, and further studies revealed that this was due to a marked depression of passive chloride absorption. Although Na+-glucose cotransport was normal in the CF jejunum, absence of passive chloride absorption completely blocked glucose-stimulated net sodium absorption and reduced glucose-stimulated water absorption 66%. This chloride absorptive abnormality acts in physiological opposition to the classic chloride secretory defect in the CF intestine. By increasing the fluidity of intraluminal contents, absence of passive chloride absorption may reduce the incidence and severity of intestinal disease in patients with CF.
Figures

References
-
- Akabas MH. Cystic fibrosis transmembrane conductance regulator: structure and function of an epithelial chloride channel. J. Biol. Chem. 2000;275:3729–3732. - PubMed
-
- Schwiebert EM, Benos DJ, Fuller CM. Cystic fibrosis: a multiple exocrinopathy caused by dysfunctions in a multifunctional transport protein. Am. J. Med. 1998;104:576–590. - PubMed
-
- Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am. J. Respir. Crit. Care Med. 1996;154:1229–1256. - PubMed
-
- Frase LL, Strickland AD, Kachel GW, Krejs GJ. Enhanced glucose absorption in the jejunum of patients with cystic fibrosis. Gastroenterology. 1985;88:478–484. - PubMed
-
- Berschneider HM, et al. Altered intestinal chloride transport in cystic fibrosis. FASEB J. 1988;2:2625–2629. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
