

New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype
- PMID: 12840783
- PMCID: PMC1180372
- DOI: 10.1086/377107
New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype
Abstract
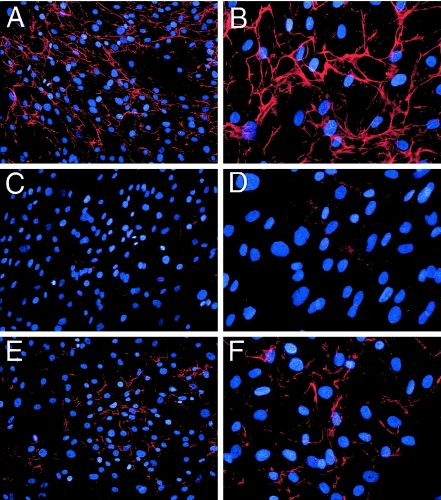
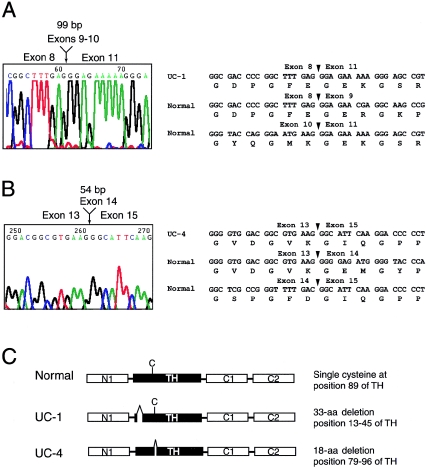
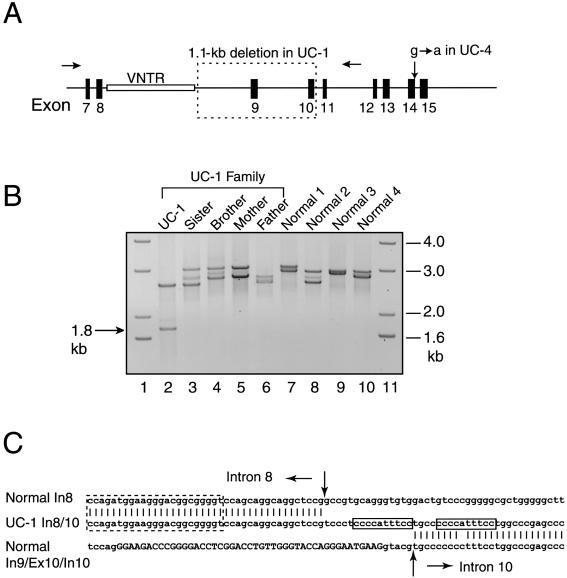
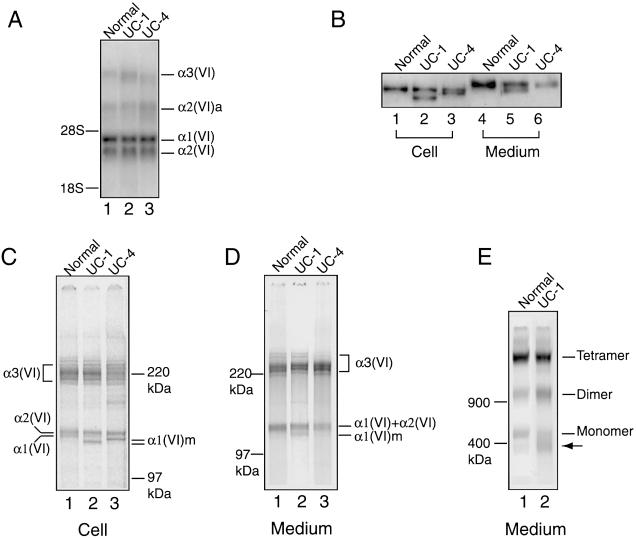
Recessive mutations in two of the three collagen VI genes, COL6A2 and COL6A3, have recently been shown to cause Ullrich congenital muscular dystrophy (UCMD), a frequently severe disorder characterized by congenital muscle weakness with joint contractures and coexisting distal joint hyperlaxity. Dominant mutations in all three collagen VI genes had previously been associated with the considerably milder Bethlem myopathy. Here we report that a de novo heterozygous deletion of the COL6A1 gene can also result in a severe phenotype of classical UCMD precluding ambulation. The internal gene deletion occurs near a minisatellite DNA sequence in intron 8 that removes 1.1 kb of genomic DNA encompassing exons 9 and 10. The resulting mutant chain contains a 33-amino acid deletion near the amino-terminus of the triple-helical domain but preserves a unique cysteine in the triple-helical domain important for dimer formation prior to secretion. Thus, dimer formation and secretion of abnormal tetramers can occur and exert a strong dominant negative effect on microfibrillar assembly, leading to a loss of normal localization of collagen VI in the basement membrane surrounding muscle fibers. Consistent with this mechanism was our analysis of a patient with a much milder phenotype, in whom we identified a previously described Bethlem myopathy heterozygous in-frame deletion of 18 amino acids somewhat downstream in the triple-helical domain, a result of exon 14 skipping in the COL6A1 gene. This deletion removes the crucial cysteine, so that dimer formation cannot occur and the abnormal molecule is not secreted, preventing the strong dominant negative effect. Our studies provide a biochemical insight into genotype-phenotype correlations in this group of disorders and establish that UCMD can be caused by dominantly acting mutations.
Figures


References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for COL6A1 [accession numbers NM_0018489 and NT_011515], COL6A2 [accession numbers NM_001849 and NT_011515], and COL6A3 [accession numbers X52022 and NT_005120])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for UCMD and Bethlem myopathy)
References
-
- Bachinger HP, Fessler LI, Timpl R, Fessler JH (1981) Chain assembly intermediate in the biosynthesis of type III procollagen in chick embryo blood vessels. J Biol Chem 256:13193–13199 - PubMed
-
- Bertini E, Pepe G (2002) Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur J Paediatr Neurol 6:193–198 - PubMed
-
- Bethlem J, Wijngaarden GK (1976) Benign myopathy, with autosomal dominant inheritance: a report on three pedigrees. Brain 99:91–100 - PubMed
-
- Camacho Vanegas O, Zhang RZ, Sabatelli P, Lattanzi G, Bencivenga P, Giusti B, Columbaro M, Chu ML, Merlini L, Pepe G (2002) Novel COL6A1 splicing mutation in a family affected by mild Bethlem myopathy. Muscle Nerve 25:513–519 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
