

doi: 10.1101/gad.1097103.
cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2
Affiliations
- PMID: 12842910
- PMCID: PMC196130
- DOI: 10.1101/gad.1097103
Item in Clipboard
cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2
Genes Dev.
.
Abstract
The incretin hormone GLP1 promotes islet-cell survival via the second messenger cAMP. Here we show that mice deficient in the activity of CREB, caused by expression of a dominant-negative A-CREB transgene in pancreatic beta-cells, develop diabetes secondary to beta-cell apoptosis. Remarkably, A-CREB severely disrupted expression of IRS2, an insulin signaling pathway component that is shown here to be a direct target for CREB action in vivo. As induction of IRS2by cAMP enhanced activation of the survival kinase Akt in response to insulin and IGF-1, our results demonstrate a novel mechanism by which opposing pathways cooperate in promoting cell survival.
Figures
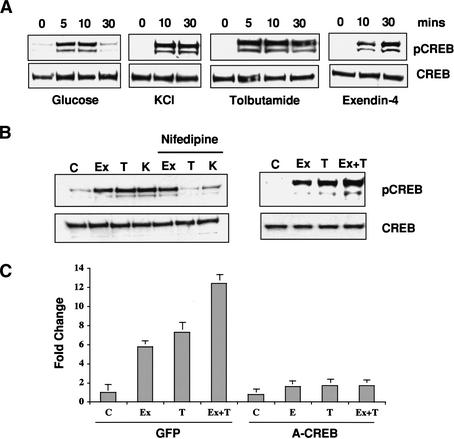
Cooperative effects of glucose and GLP-1 signaling on CREB activation.
(A) Western blot assay of phospho (Ser 133) and total CREB levels in
MIN6 cells following stimulation with high glucose, KCl depolarization,
KATP channel antagonist tolbutamide, or GLP1 agonist exendin-4.
(B, left) Effect of L-type calcium channel antagonist nifedipine on
CREB phosphorylation in MIN6 cells following treatment (30 min) with exendin-4
(Ex), tolbutamide (T), and KCl (K). (Right) Additive effect of
glucose and GLP1 signaling on CREB phosphorylation. Cells were treated with
the GLP-1 analog exendin-4 and tolbutamide, either individually or together,
and analyzed for CREB Ser 133 phosphorylation. (C) Quantitative PCR
analysis of endogenous CREB target gene (NR4A2) expression in response to
glucose, tolbutamide, and exendin-4, individually and in combination. Effect
of dominant-negative CREB (A-CREB) adenovirus or control (GFP) adenovirus
infection on NR4A2 expression.
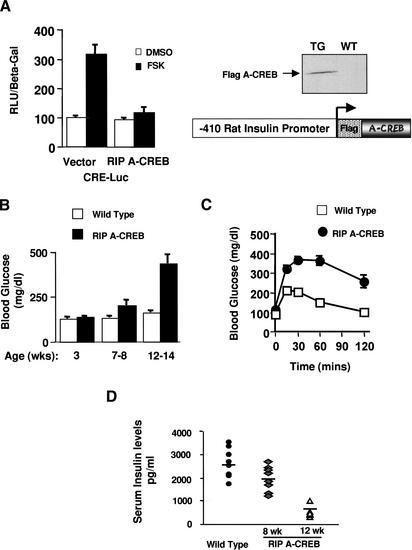
RIP A-CREB transgenic mice develop diabetes. (A) Schematic and
transient transfection assay of the RIP A-CREB expression vector in HIT-T15
insulinoma cells using the somatostatin CRE luciferase reporter.
(Right) Western blot of islet lysates from wild-type and RIP A-CREB
mice using anti-Flag tag antiserum. (B) Ambient glucose levels in RIP
A-CREB transgenic mice (n = 15) compared with wild-type littermates
(n = 12) at 3, 7–8, or 12–14 wk of age. (C)
Glucose tolerance curves of RIP A-CREB and wild-type mice at 8 wk. Glucose
levels were monitored at the times indicated after intraperitoneal injection
(2 g/kg) of glucose. (D) Serum insulin levels in wild-type (2694
± 205 pg/mL; n = 8) compared with A-CREB transgenic mice at 8
wk (1930 ± 211 pg/mL; n = 7), and 12 wk (462 ± 48
pg/mL; n = 8).
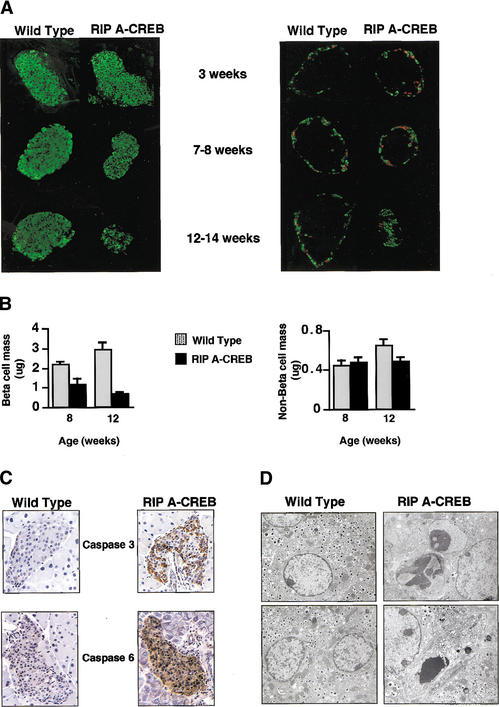
Progressive loss of islet-cell mass and cellular apoptosis in RIP A-CREB
transgenic mice. (A) Immunohistochemical staining of pancreatic
sections from transgenic and control mice at 3, 8, and 12 wk of age using
anti-insulin (green) antibodies (left) or somatostatin (green) and
glucagon (red) antisera (right). (B) Bar graph showing
relative mass in transgenic and control mice by morphometric analysis.
(C) Immunocytochemical staining for activated caspases in pancreatic
sections of wild-type and A-CREB transgenic littermates. Staining for
caspase-3 performed on 5-week-old mice, and staining for caspase-6 performed
on 10-week-old mice. (D) Electron microscopic analysis of islets from
wild-type and A-CREB transgenic mice. (Left) Normal β-cells with
evenly distributed heterochromatin and characteristic insulin granules
(magnification 11,360×). (Right, top) Islet β-cells from
A-CREB mice in early stages of apoptosis with redistribution of chromatin into
electron dense aggregates (magnification 14,560×). (Right,
bottom) End-stage apoptosis (magnification 11,360×) characterized
by progressive cytoplasmic vacuolation and nuclear fragmentation.
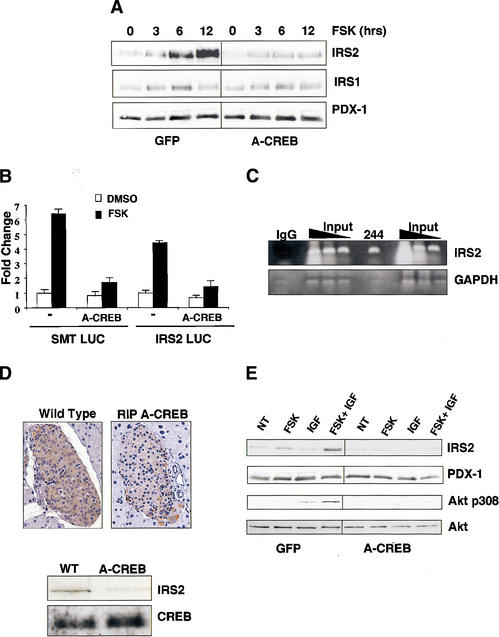
CREB promotes insulin/IGF signaling via induction of IRS2. (A)
Western blot analysis of IRS1 and IRS2 levels in MIN-6 cells treated with
forskolin for increasing times in serum-supplemented medium. Cells were
infected with A-CREB or control GFP adenovirus as indicated. (B)
Transient transfection assay of 293T cells with IRS2 promoter construct or
control somatostatin CRE luciferase plasmid. Cells treated with forskolin or
DMSO vehicle. Cotransfection with A-CREB expression plasmid is indicated.
(C) Chromatin immunoprecipitation assay of the IRS2 promoter
using CREB 244 antiserum. Presence of the IRS2 promoter or negative
control GAPDH in CREB immunoprecipitates determined by PCR amplification of
anti-CREB or control IgG immunoprecipitates. (D) IRS2 expression is
disrupted in A-CREB transgenic mice. (Top) Immunohistochemical
staining of pancreatic sections from wild-type and RIP A-CREB transgenic mice
using anti-IRS2 antiserum. (Bottom) Western blot assay of islet-cell
extracts from wild-type and A-CREB mice using anti-IRS2 antiserum. Comparable
levels of CREB protein in transgenic and wild-type lysates. (E)
cAMP-dependent induction of IRS-2 potentiates growth factor signaling to Akt.
Effect of IGF1 on Akt phosphorylation at Thr 308 in cells pretreated with
forskolin or vehicle under serum-starved conditions for 8 h. Effect of A-CREB
on IGF-stimulated Akt Thr 308 phosphorylation.
References
-
- Bonner-Weir S., Deery, D., Leahy, J.L., and Weir, G.C. 1989. Compensatory growth of pancreatic β-cells in adult rats after short-term glucose infusion. Diabetes 38: 49–53. - PubMed
-
- Bruning J.C., Winnay, J., Bonner-Weir, S., Taylor, S.I., Accili, D., and Kahn, C.R. 1997. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 88: 561–572. - PubMed
-
- Canettieri G., Morantte, I., Guzman, E., Asahara, H., Herzig, S., Anderson, S.D., Yates, J.R., and Montminy, M. 2003. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat. Struct. Biol. 10: 175–181. - PubMed
-
- Conkright M.D., Guzmán, E., Flechner, L., Su, A.I., Hogenesch, J., and Montminy, M. 2003. Genome wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol. Cell 11: 1101–1108. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous