

Novel role of vitamin k in preventing oxidative injury to developing oligodendrocytes and neurons
- PMID: 12843286
- PMCID: PMC6741273
- DOI: 10.1523/JNEUROSCI.23-13-05816.2003
Novel role of vitamin k in preventing oxidative injury to developing oligodendrocytes and neurons
Abstract
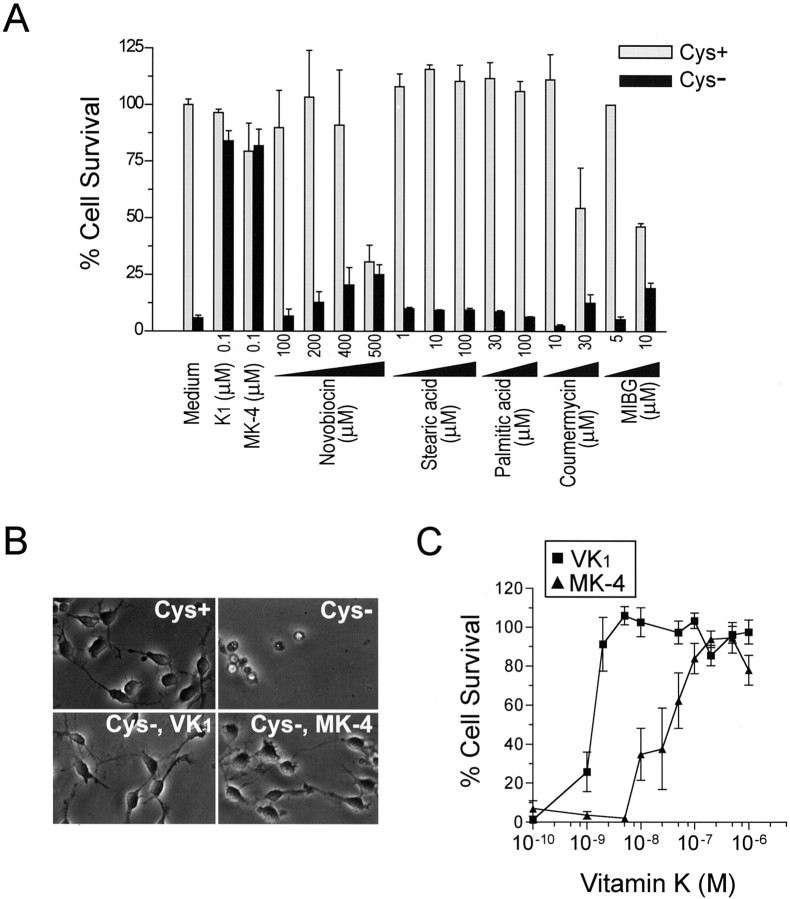
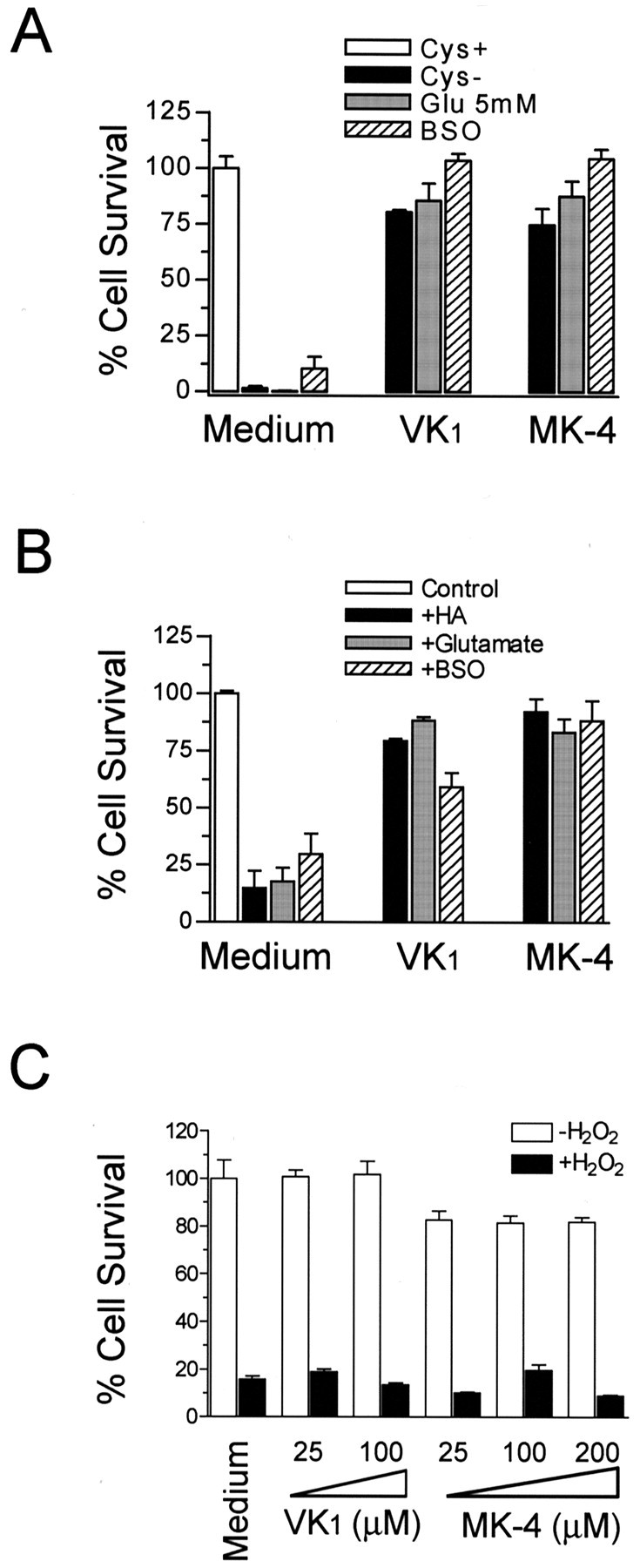
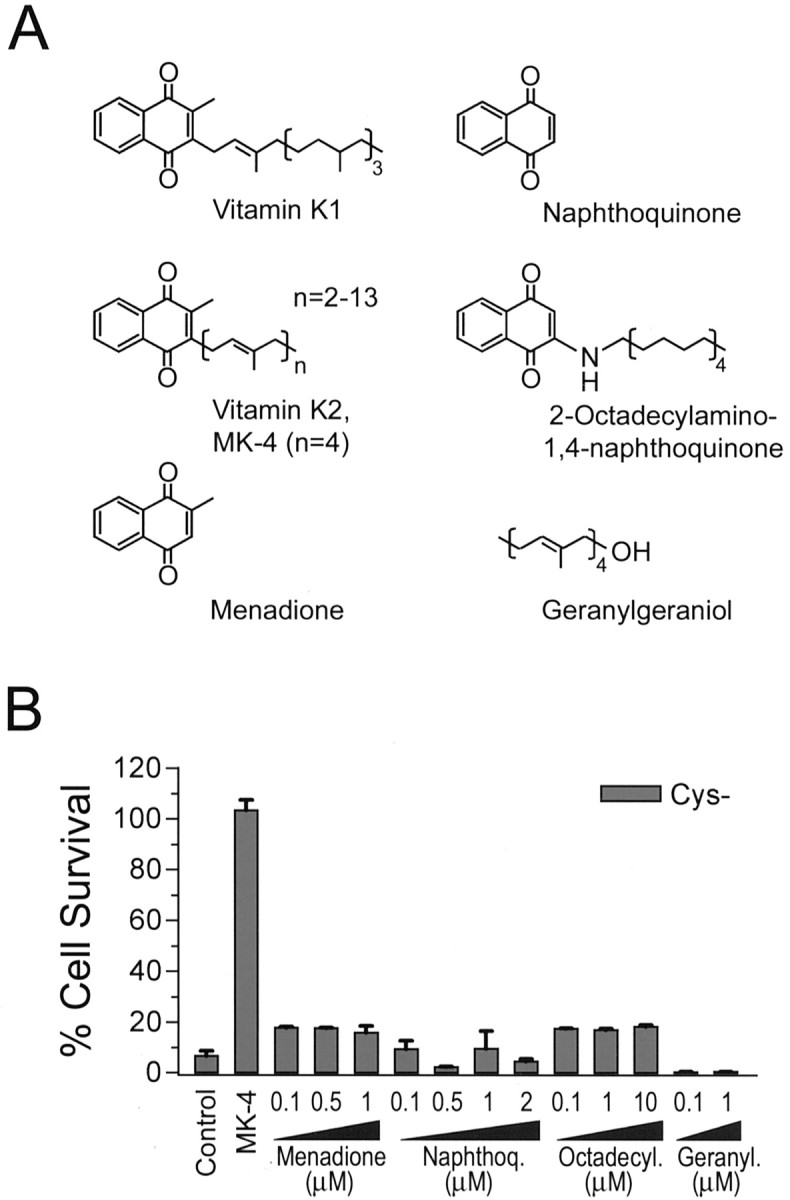
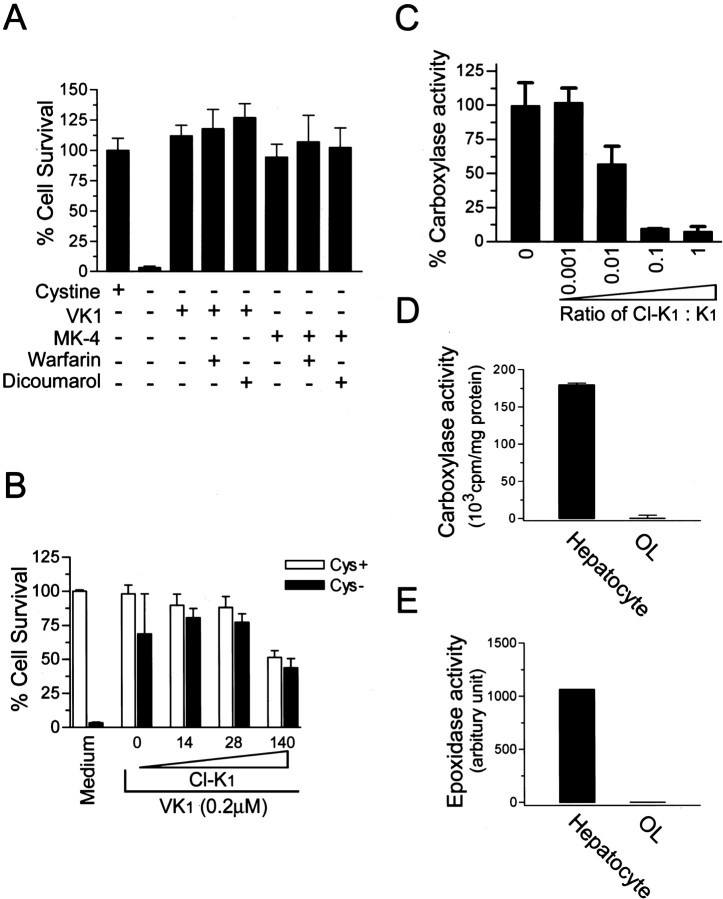
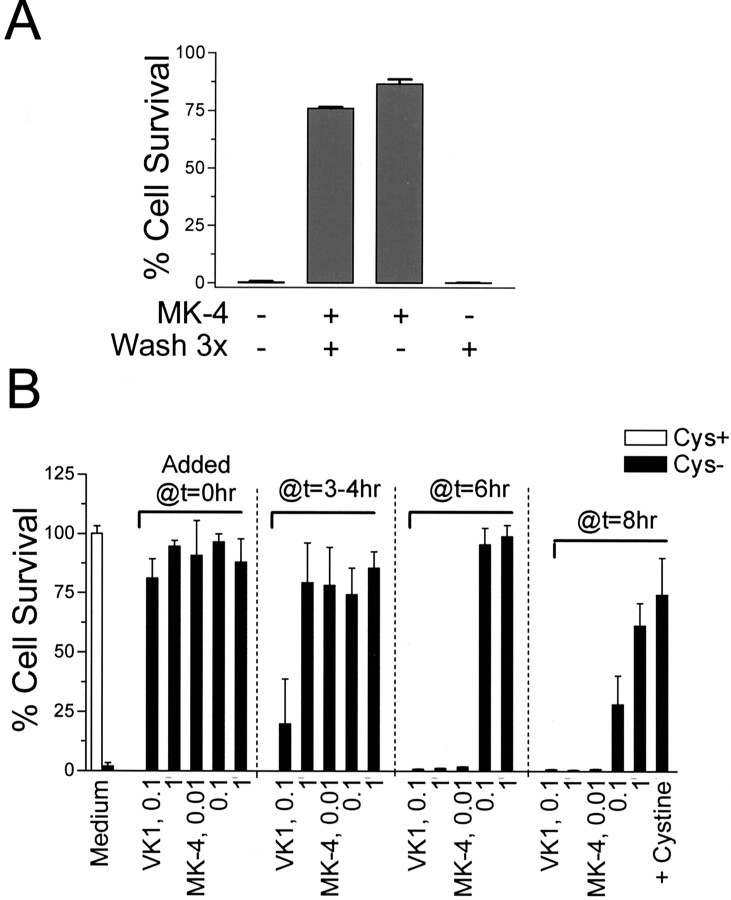
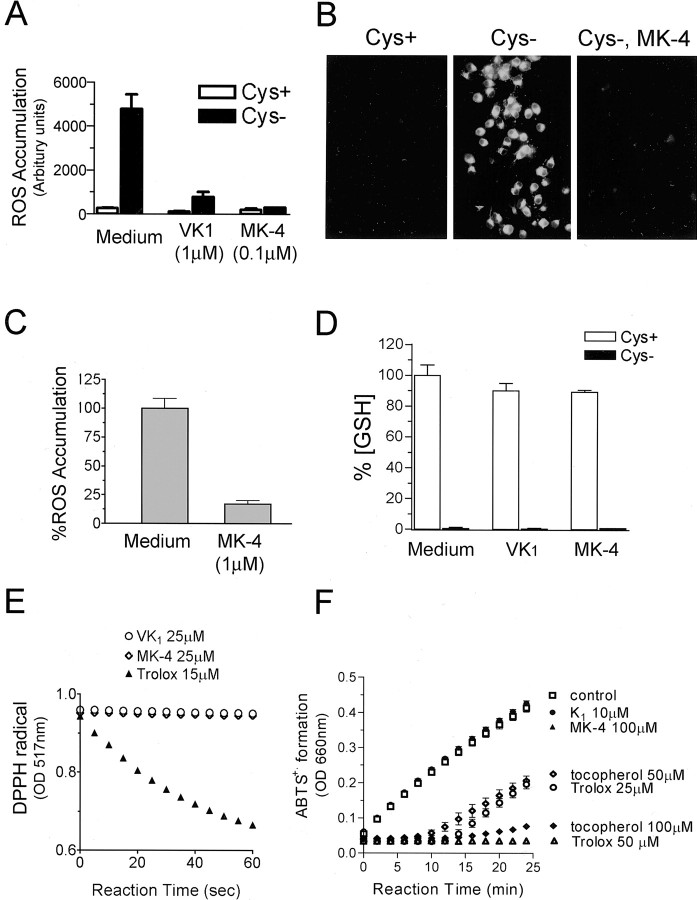
Oxidative stress is believed to be the cause of cell death in multiple disorders of the brain, including perinatal hypoxia/ischemia. Glutamate, cystine deprivation, homocysteic acid, and the glutathione synthesis inhibitor buthionine sulfoximine all cause oxidative injury to immature neurons and oligodendrocytes by depleting intracellular glutathione. Although vitamin K is not a classical antioxidant, we report here the novel finding that vitamin K1 and K2 (menaquinone-4) potently inhibit glutathione depletion-mediated oxidative cell death in primary cultures of oligodendrocyte precursors and immature fetal cortical neurons with EC50 values of 30 nm and 2 nm, respectively. The mechanism by which vitamin K blocks oxidative injury is independent of its only known biological function as a cofactor for gamma-glutamylcarboxylase, an enzyme responsible for posttranslational modification of specific proteins. Neither oligodendrocytes nor neurons possess significant vitamin K-dependent carboxylase or epoxidase activity. Furthermore, the vitamin K antagonists warfarin and dicoumarol and the direct carboxylase inhibitor 2-chloro-vitamin K1 have no effect on the protective function of vitamin K against oxidative injury. Vitamin K does not prevent the depletion of intracellular glutathione caused by cystine deprivation but completely blocks free radical accumulation and cell death. The protective and potent efficacy of this naturally occurring vitamin, with no established clinical side effects, suggests a potential therapeutic application in preventing oxidative damage to undifferentiated oligodendrocytes in perinatal hypoxic/ischemic brain injury.
Figures
References
-
- Back SA, Khan R, Gan X, Rosenberg PA, Volpe JJ ( 1999) A new Alamar Blue viability assay to rapidly quantify oligodendrocyte death. J Neurosci Methods 91: 47-54. - PubMed
-
- Banasik M, Komura H, Shimoyama M, Ueda K ( 1992) Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl)transferase. J Biol Chem 267: 1569-1575. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources