

14-3-3s regulate fructose-2,6-bisphosphate levels by binding to PKB-phosphorylated cardiac fructose-2,6-bisphosphate kinase/phosphatase
- PMID: 12853467
- PMCID: PMC165633
- DOI: 10.1093/emboj/cdg363
14-3-3s regulate fructose-2,6-bisphosphate levels by binding to PKB-phosphorylated cardiac fructose-2,6-bisphosphate kinase/phosphatase
Abstract
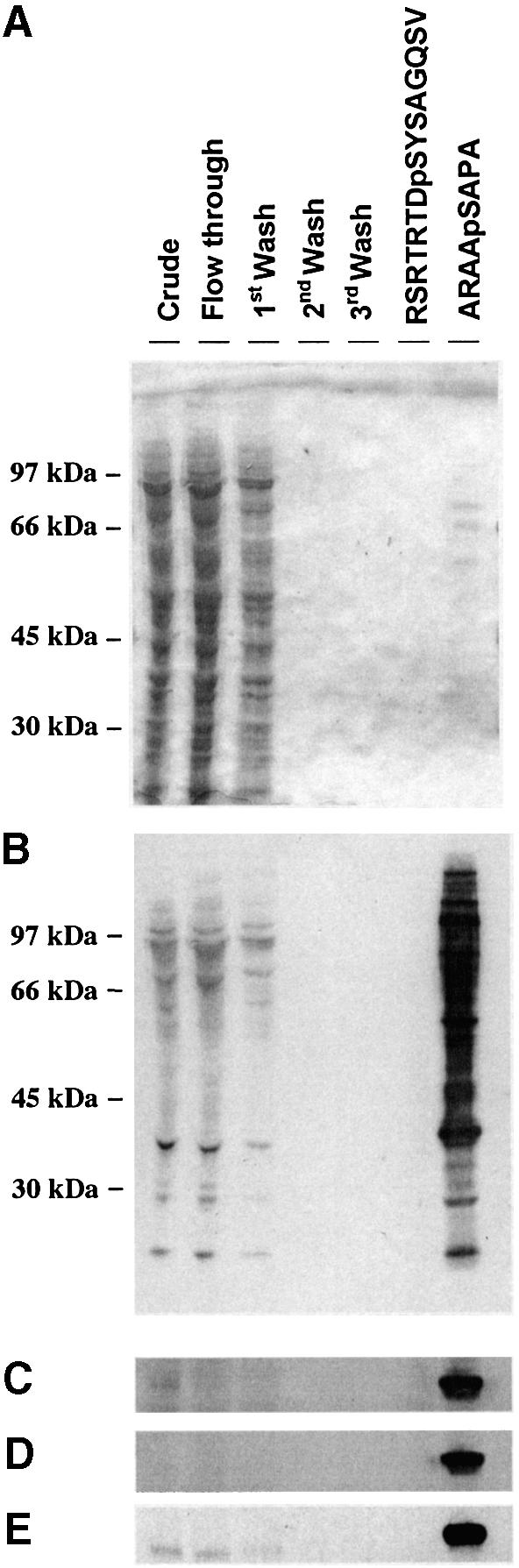
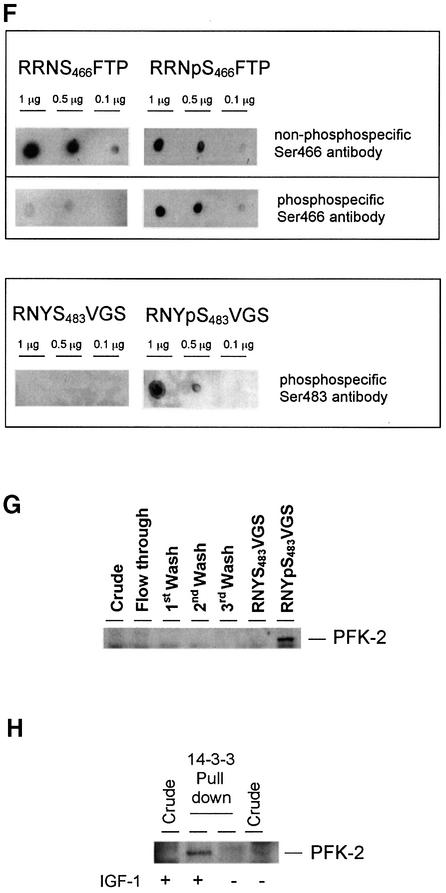
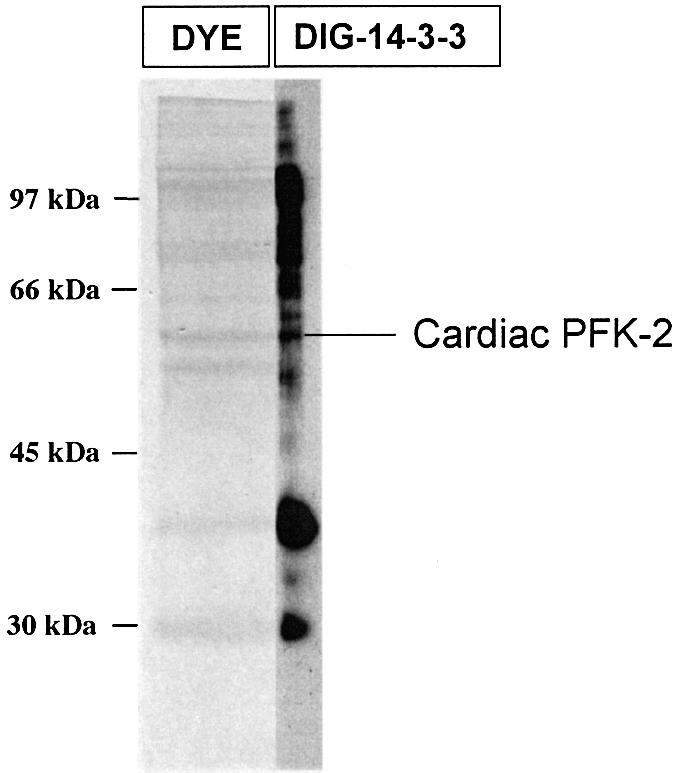
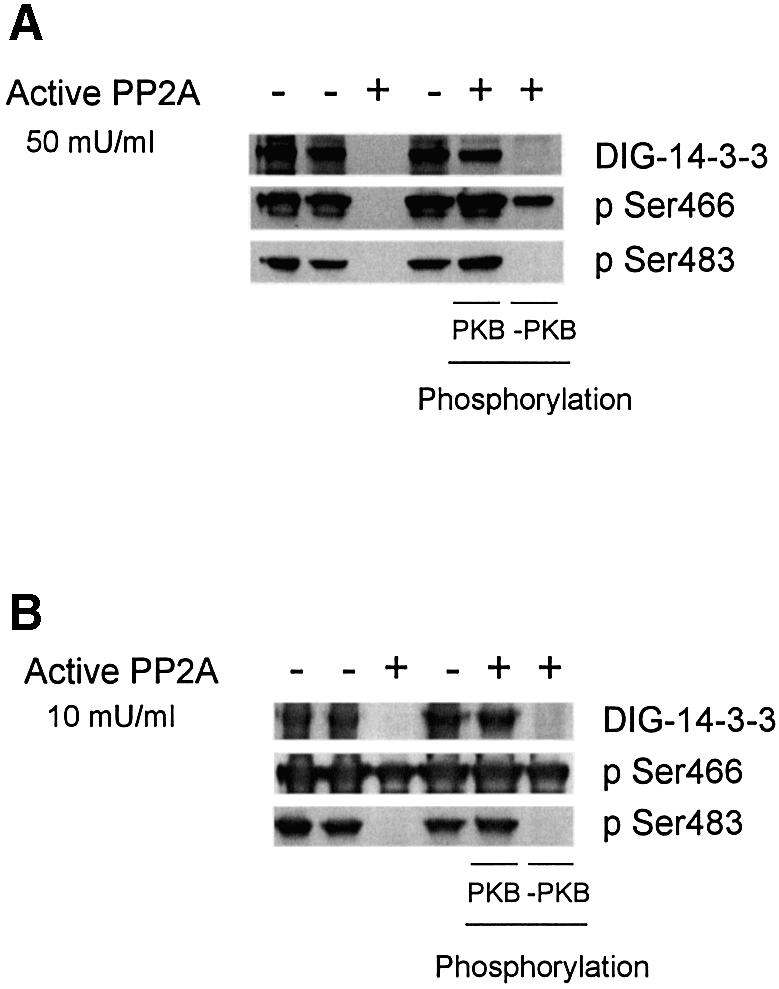
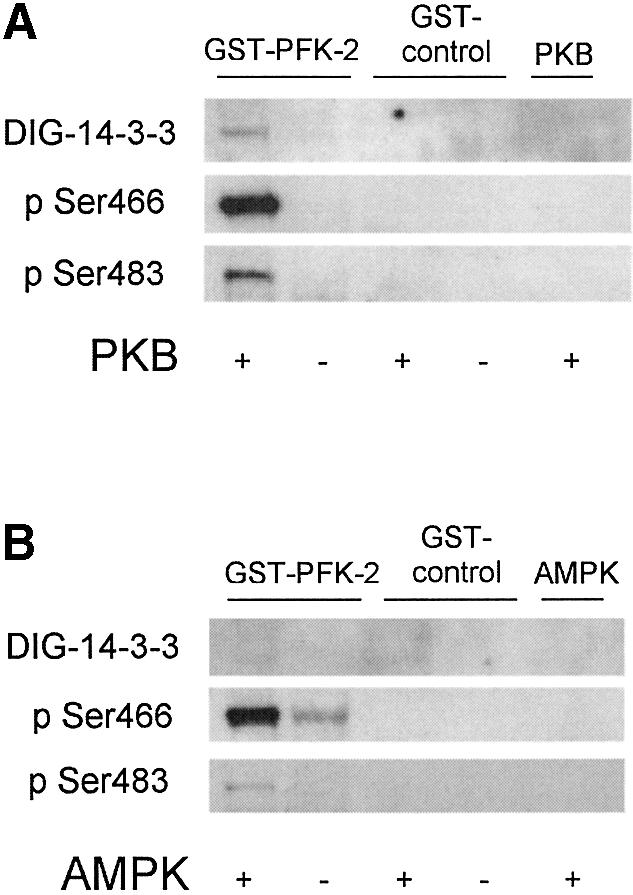
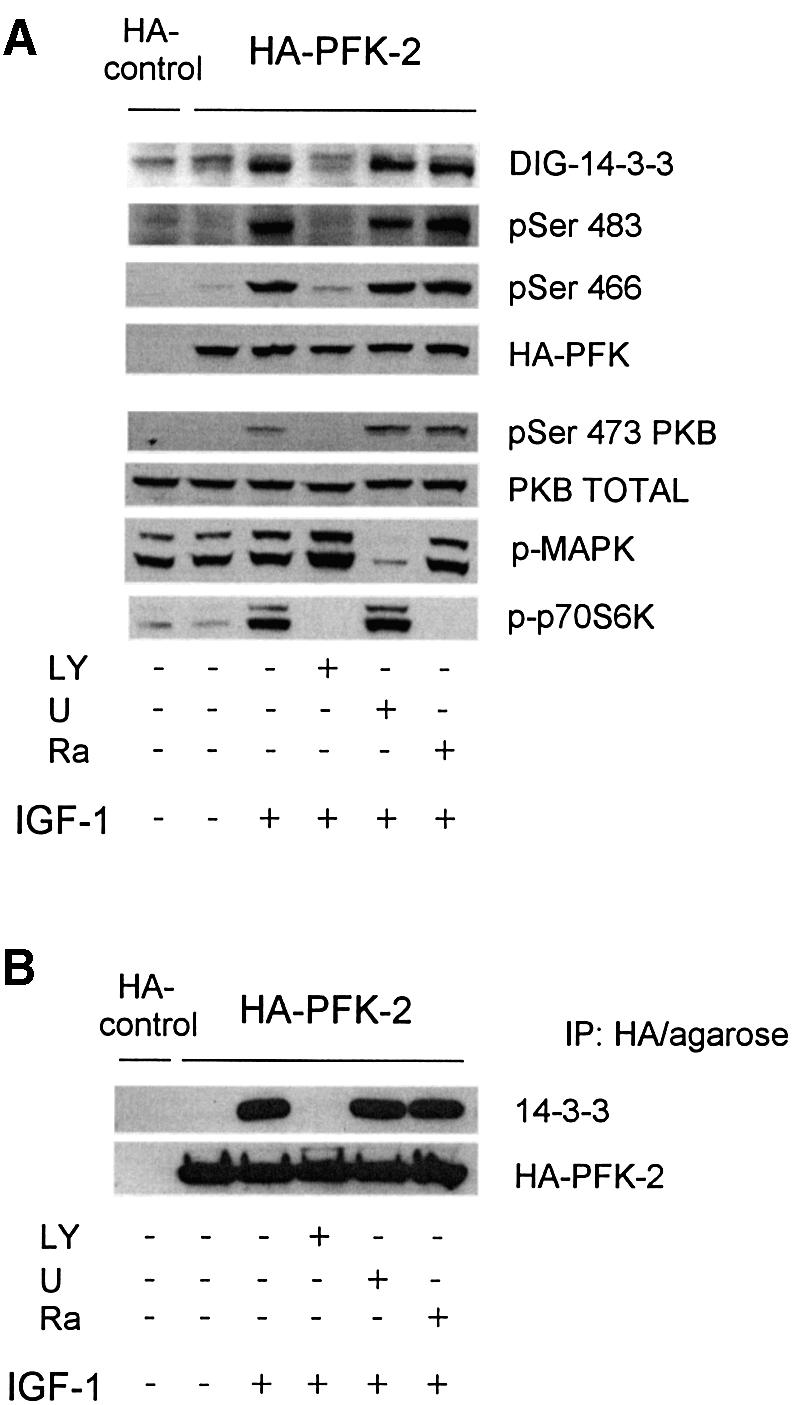
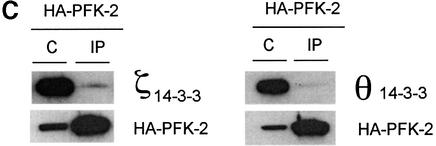
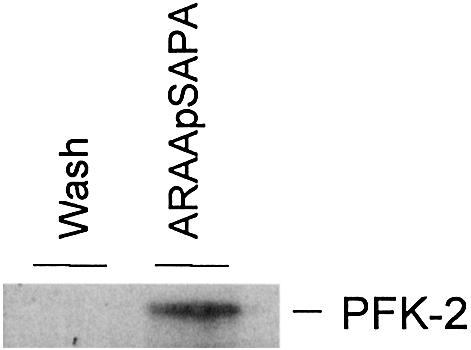
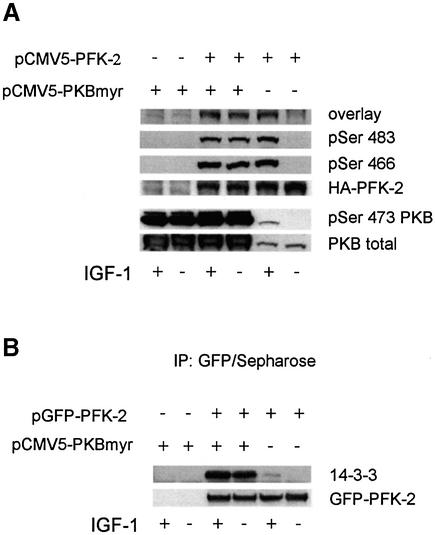
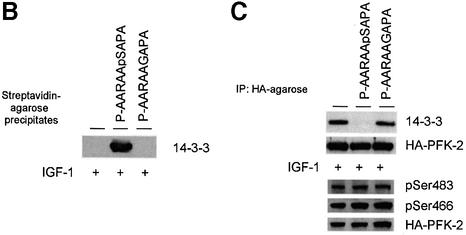
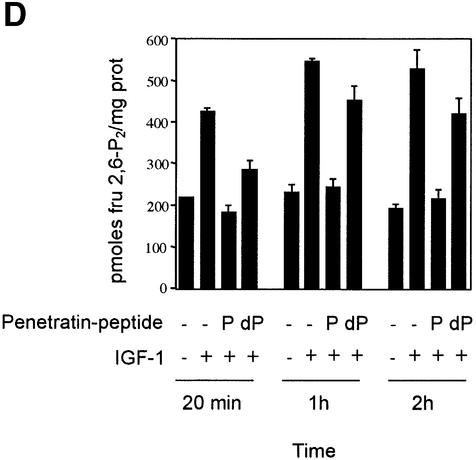
The cardiac isoform of 6-phosphofructo-2-kinase/ fructose-2,6-bisphosphatase (PFK-2), regulator of the glycolysis-stimulating fructose-2,6-bisphosphate, was among human HeLa cell proteins that were eluted from a 14-3-3 affinity column using the phosphopeptide ARAApSAPA. Tryptic mass fingerprinting and phospho-specific antibodies showed that Ser466 and Ser483 of 14-3-3-affinity-purified PFK-2 were phosphorylated. 14-3-3 binding was abolished by selectively dephosphorylating Ser483, and 14-3-3 binding was restored when both Ser466 and Ser483 were phosphorylated with PKB, but not when Ser466 alone was phosphorylated by AMPK. Furthermore, the phosphopeptide RNYpS(483)VGS blocked binding of PFK-2 to 14-3-3s. These data indicate that 14-3-3s bind to phosphorylated Ser483. When HeLa cells expressing HA-tagged PFK-2 were co-transfected with active PKB or stimulated with IGF-1, HA-PFK-2 was phosphorylated and bound to 14-3-3s. The response to IGF-1 was abolished by PI 3-kinase inhibitors. In addition, IGF-1 promoted the binding of endogenous PFK-2 to 14-3-3s. When cells were transduced with penetratin-linked AARAApSAPA, we found that this reagent bound specifically to 14-3-3s, blocked the IGF-1-induced binding of HA-PFK-2 to 14-3-3s, and completely inhibited the IGF-1-induced increase in cellular fructose-2,6-bisphosphate. These findings suggest that PKB-dependent binding of 14-3-3s to phospho-Ser483 of cardiac PFK-2 mediates the stimulation of glycolysis by growth factor.
Figures

References
-
- Andjelkovic M. et al. (1997) Role of translocation in the activation and function of protein kinase B. J. Biol. Chem., 272, 31515–31524. - PubMed
-
- Bertrand L., Alessi,D.R., Deprez,J., Deak,M., Viaene,E., Rider,M.H. and Hue,L. (1999) Heart 6-phosphofructo-2-kinase activation by insulin results from Ser-466 and Ser-483 phosphorylation and requires 3-phosphoinositide-dependent kinase-1, but not protein kinase B. J. Biol. Chem., 274, 30927–30933. - PubMed
-
- Chesney J., Mitchell,R., Benigni,F., Bacher,M., Spiegel,L., Al-Abed,Y., Han,J.H., Metz,C. and Bucala,R. (1999) An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect. Proc. Natl Acad. Sci. USA, 96, 3047–3052. - PMC - PubMed
-
- Dale S., Wilson,W.A., Edelman,A.M. and Hardie,D.G. (1995) Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1 and mammalian calmodulin-dependent protein kinase I. FEBS Lett., 361, 191–195. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
