

Malignancy-associated regions of transcriptional activation: gene expression profiling identifies common chromosomal regions of a recurrent transcriptional activation in human prostate, breast, ovarian, and colon cancers
- PMID: 12869305
- PMCID: PMC1502409
- DOI: 10.1016/S1476-5586(03)80054-4
Malignancy-associated regions of transcriptional activation: gene expression profiling identifies common chromosomal regions of a recurrent transcriptional activation in human prostate, breast, ovarian, and colon cancers
Abstract
Despite remarkable advances in our understanding of a genetic basis of cancer, the precise molecular definition of the phenotypically relevant genetic features associated with human epithelial malignancies remains a significant and highly relevant challenge. Here we performed a systematic analysis of the chromosomal positions of cancer-associated transcripts for prostate, breast, ovarian, and colon tumors, and identified short segments of human chromosomes that appear to represent a common target for transcriptional activation in major epithelial malignancies in human. These cancer-associated transcriptomeres correspond well to the regions of transient transcriptional activity on chromosomes 1q21-q23 (144-160 Mbp), 12q13 (52-63 Mbp), 17q21 (38-50 Mbp), 17q23-q25 (72-82 Mbp), 19p13 (1-16 Mbp), and Xq28 (132-142 Mbp) during human cell cycle, suggesting a common epigenetic mechanism of transcriptional activation. Consistent with this idea, two of these transcriptomeres (12q13 and 17q21) seemed to be related to the p53-regulated transcriptional clusters, and some of the cancer-associated transcriptomeres appeared to correspond well to the recently identified regions of increased gene expression on human chromosomes.
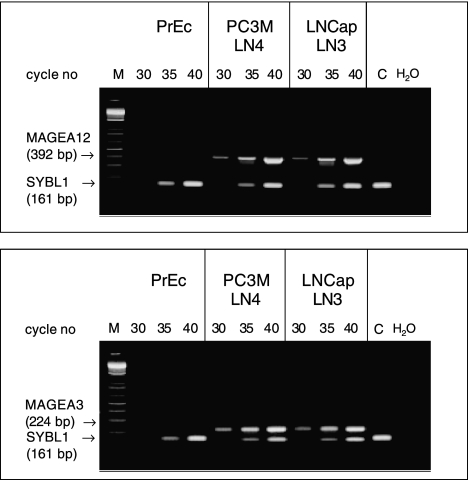
Figures





Similar articles
-
Common malignancy-associated regions of transcriptional activation (MARTA) in human prostate, breast, ovarian, and colon cancers are targets for DNA amplification.Cancer Lett. 2003 Nov 10;201(1):67-77. doi: 10.1016/s0304-3835(03)00419-1. Cancer Lett. 2003. PMID: 14580688
-
Expression profiling targeting chromosomes for tumor classification and prediction of clinical behavior.Genes Chromosomes Cancer. 2003 Nov;38(3):207-14. doi: 10.1002/gcc.10276. Genes Chromosomes Cancer. 2003. PMID: 14506694
-
Different mechanisms are implicated in ERBB2 gene overexpression in breast and in other cancers.Br J Cancer. 2003 Sep 1;89(5):899-906. doi: 10.1038/sj.bjc.6601200. Br J Cancer. 2003. PMID: 12942124 Free PMC article.
-
Involvement of the multiple tumor suppressor genes and 12-lipoxygenase in human prostate cancer. Therapeutic implications.Adv Exp Med Biol. 1997;407:41-53. doi: 10.1007/978-1-4899-1813-0_7. Adv Exp Med Biol. 1997. PMID: 9321930 Review.
-
BRCA1 and prostate cancer.Cancer Invest. 2001;19(4):396-412. doi: 10.1081/cnv-100103134. Cancer Invest. 2001. PMID: 11405179 Review.
Cited by
-
A review of the past, present, and future directions of neoplasia.Neoplasia. 2005 Dec;7(12):1039-46. doi: 10.1593/neo.05793. Neoplasia. 2005. PMID: 16354585 Free PMC article. Review. No abstract available.
-
In-silico identification and functional validation of allele-dependent AR enhancers.Oncotarget. 2015 Mar 10;6(7):4816-28. doi: 10.18632/oncotarget.3019. Oncotarget. 2015. PMID: 25693204 Free PMC article.
-
mRNA profiling of the cancer degradome in oesophago-gastric adenocarcinoma.Br J Cancer. 2012 Jun 26;107(1):143-9. doi: 10.1038/bjc.2012.239. Epub 2012 Jun 7. Br J Cancer. 2012. PMID: 22677901 Free PMC article.
-
Predicting the clinical course of prostate cancer.J Clin Invest. 2004 Mar;113(6):806-8. doi: 10.1172/JCI21310. J Clin Invest. 2004. PMID: 15067311 Free PMC article.
-
Topoisomerase II inhibition involves characteristic chromosomal expression patterns.BMC Genomics. 2008 Jul 8;9:324. doi: 10.1186/1471-2164-9-324. BMC Genomics. 2008. PMID: 18611269 Free PMC article.
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Welsh J, Jeffrey SS, Botstein D, Brown PO. Genome-wide analysis of DNA-copy number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. - PubMed
-
- Forozan F, Mahlamaki EH, Monni O, Chen Y, Veldman R, Jiang Y, Gooden GC, Ethier SP, Kallioniemi A, Kallioniemi O-P. Comparative genomic hybridization analysis of 38 breast cancer cell lines: a basis for interpreting complementary DNA microarray data. Cancer Res. 2000;60:4519–4525. - PubMed
-
- Caron H, van Schaik B, van der Mee M, Baas F, Riggins G, van Stuis P, Hermus M-C, van Asperen R, Boon K, Voute PA, Heisterkamp S, van Kampen A, Versteeg R. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–1292. - PubMed
-
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portrait of human breast tumors. Nature. 2000;406:747–752. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
