

Structure-based phenotyping predicts HIV-1 protease inhibitor resistance
- PMID: 12876320
- PMCID: PMC2323957
- DOI: 10.1110/ps.0301103
Structure-based phenotyping predicts HIV-1 protease inhibitor resistance
Abstract
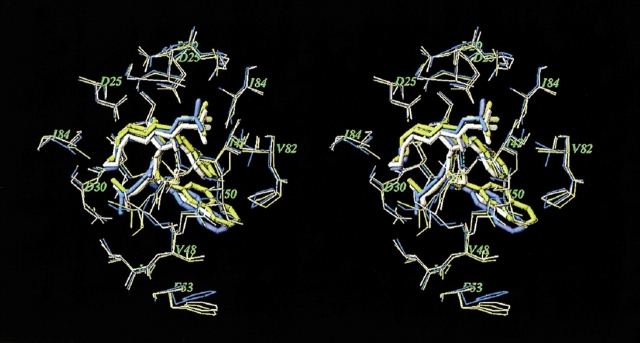
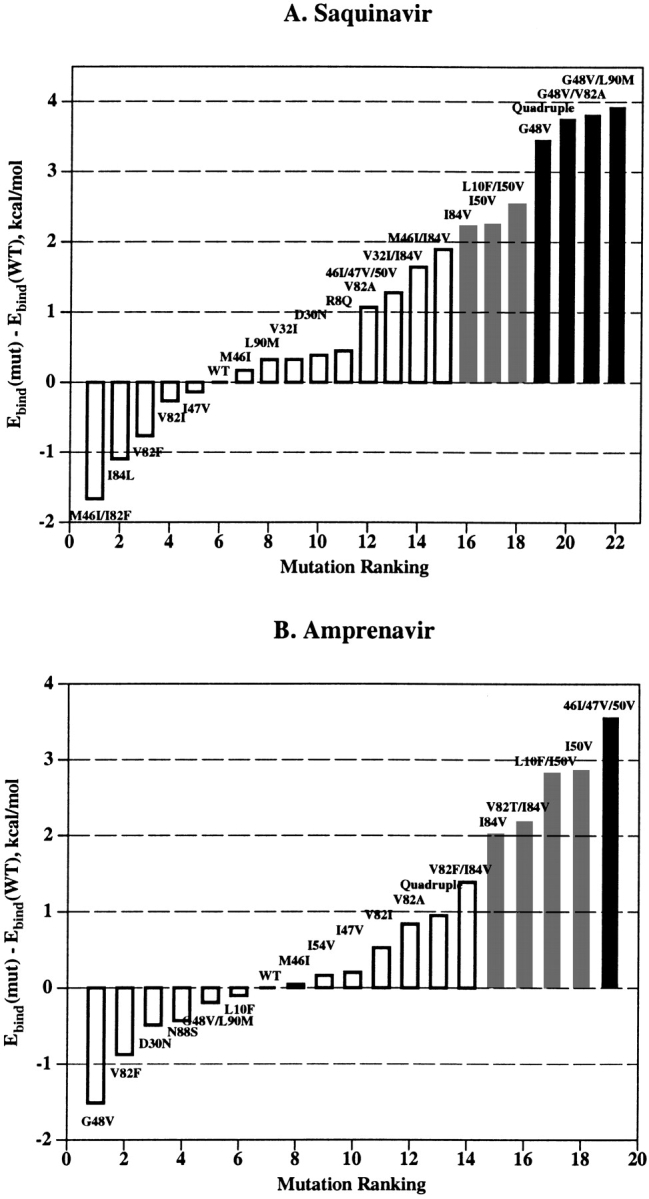
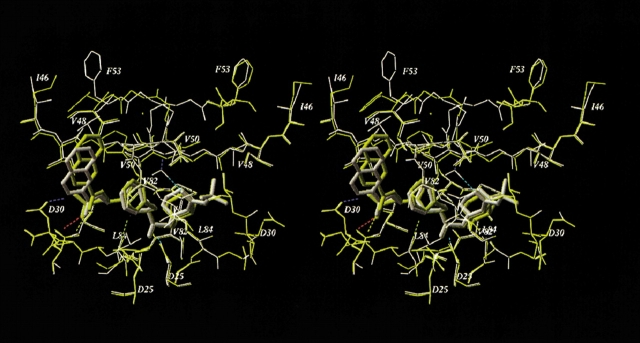
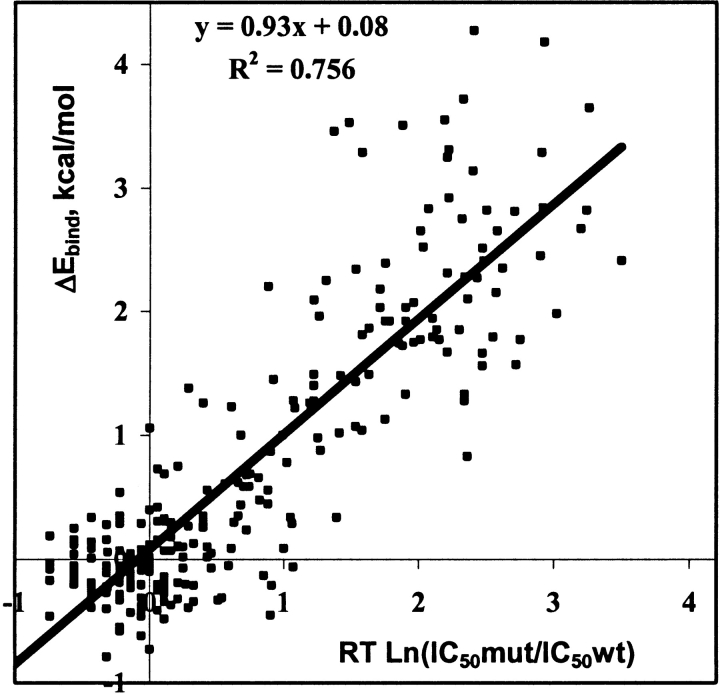
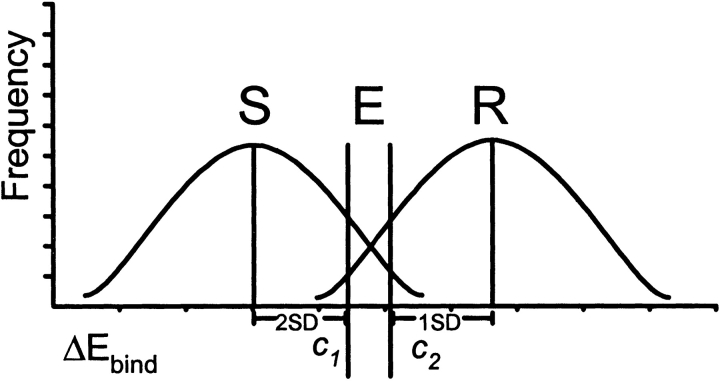
Mutations in HIV-1 drug targets lead to resistance and consequent therapeutic failure of antiretroviral drugs. Phenotypic resistance assays are time-consuming and costly, and genotypic rules-based interpretations may fail to predict the effects of multiple mutations. We have developed a computational procedure that rapidly evaluates changes in the binding energy of inhibitors to mutant HIV-1 PR variants. Models of WT complexes were produced from crystal structures. Mutant complexes were built by amino acid substitutions in the WT complexes with subsequent energy minimization of the ligand and PR binding site residues. Accuracy of the models was confirmed by comparison with available crystal structures and by prediction of known resistance-related mutations. PR variants from clinical isolates were modeled in complex with six FDA-approved PIs, and changes in the binding energy (DeltaE(bind)) of mutant versus WT complexes were correlated with the ratios of phenotypic 50% inhibitory concentration (IC(50)) values. The calculated DeltaE(bind) of five PIs showed significant correlations (R(2) = 0.7-0.8) with IC(50) ratios from the Virco Antivirogram assay, and the DeltaE(bind) of six PIs showed good correlation (R(2) = 0.76-0.85) with IC(50) ratios from the Virologic PhenoSense assay. DeltaE(bind) cutoffs corresponding to a four-fold increase in IC(50) were used to define the structure-based phenotype as susceptible, resistant, or equivocal. Blind predictions for 78 PR variants gave overall agreement of 92% (kappa = 0.756) and 86% (kappa = 0.666) with PhenoSense and Antivirogram phenotypes, respectively. The structural phenotyping predicted drug resistance of clinical HIV-1 PR variants with an accuracy approaching that of frequently used cell-based phenotypic assays.
Figures
References
-
- Abagyan, R. and Totrov, M. 1994. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235 983–1002. - PubMed
-
- Abagyan, R.A., Totrov, M., and Kuznetsov, D. 1994. ICM—a new method for protein modeling and design: Applications to docking and structure prediction from the disordered native conformation. J. Comput. Chem. 15 488–506.
-
- Baldwin, E.T., Bhat, T.N., Liu, B., Pattabiraman, N., and Erickson, J.W. 1995. Structural basis of drug resistance for the V82A mutant of HIV-1 proteinase. Nat. Struct. Biol. 2 244–249. - PubMed
-
- Baxter, J.D., Mayers, D.L., Wentworth, D.N., Neaton, J.D., Hoover, M.L., Winters, M.A., Mannheimer, S.B., Thompson, M.A., Abrams, D.I., Brizz, B.J., et al. 2000. A randomized study of antiretroviral management based on plasma genotypic retroviral resistance testing in patients failing therapy. AIDS 14 F83–93. - PubMed
-
- Carpenter, C.C., Cooper, D.A., Fischl, M.A., Gatell, J.M., Gazzard, B.G., Hammer, S.M., Hirsch, M.S., Jacobsen, D.M., Katzenstein, D.A., Montaner, J.S., et al. 2000. Antiretroviral therapy in adults: Updated recommendations of the International AIDS Society-USA Panel. JAMA 283 381–390. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
