

An activated receptor tyrosine kinase, TEL/PDGFbetaR, cooperates with AML1/ETO to induce acute myeloid leukemia in mice
- PMID: 12881486
- PMCID: PMC170948
- DOI: 10.1073/pnas.1531730100
An activated receptor tyrosine kinase, TEL/PDGFbetaR, cooperates with AML1/ETO to induce acute myeloid leukemia in mice
Abstract
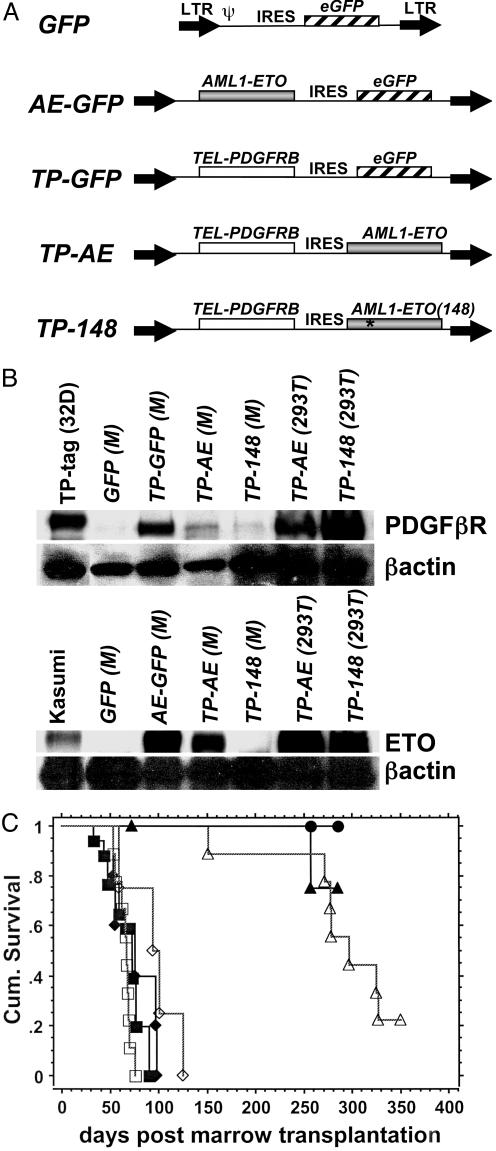
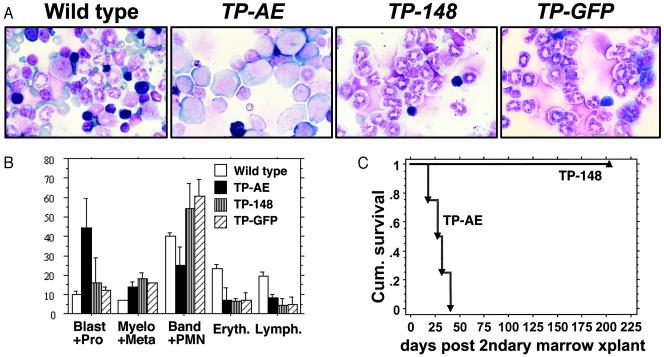
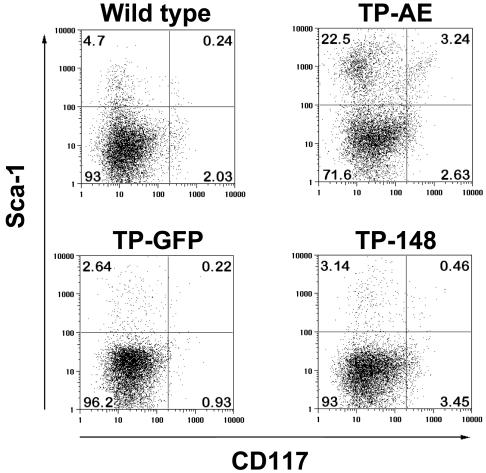
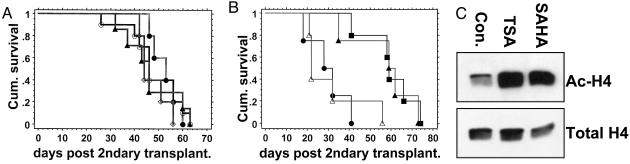
The t(8;21)(q22;q22) translocation, occurring in 40% of patients with acute myeloid leukemia (AML) of the FAB-M2 subtype (AML with maturation), results in expression of the RUNX1-CBF2T1 [AML1-ETO (AE)] fusion oncogene. AML/ETO may contribute to leukemogenesis by interacting with nuclear corepressor complexes that include histone deacetylases, which mediate the repression of target genes. However, expression of AE is not sufficient to transform primary hematopoietic cells or cause disease in animals, suggesting that additional mutations are required. Activating mutations in receptor tyrosine kinases (RTK) are present in at least 30% of patients with AML. To test the hypothesis that activating RTK mutations cooperate with AE to cause leukemia, we transplanted retrovirally transduced murine bone marrow coexpressing TEL-PDGFRB and AE into lethally irradiated syngeneic mice. These mice (19/19, 100%) developed AML resembling M2-AML that was transplantable in secondary recipients. In contrast, control mice coexpressing with TEL-PDGFRB and a DNA-binding-mutant of AE developed a nontransplantable myeloproliferative disease identical to that induced by TEL-PDGFRB alone. We used this unique model of AML to test the efficacy of pharmacological inhibition of histone deacetylase activity by using trichostatin A and suberoylanilide hydroxamic acid alone or in combination with the tyrosine kinase inhibitor, imatinib mesylate. We found that although imatinib prolonged the survival of treated mice, histone deacetylase inhibitors provided no additional survival benefit. These data demonstrate that an activated RTK can cooperate with AE to cause AML in mice, and that this system can be used to evaluate novel therapeutic strategies.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
