

Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1)
- PMID: 12897212
- PMCID: PMC166299
- DOI: 10.1172/JCI18264
Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1)
Erratum in
- J Clin Invest. 2005 Apr;115(4):1100
Abstract
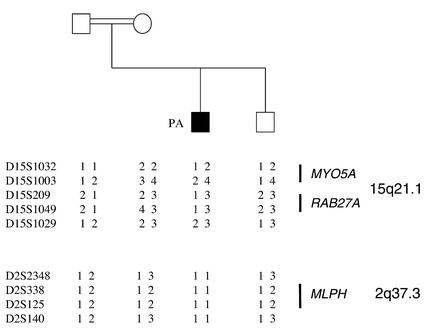
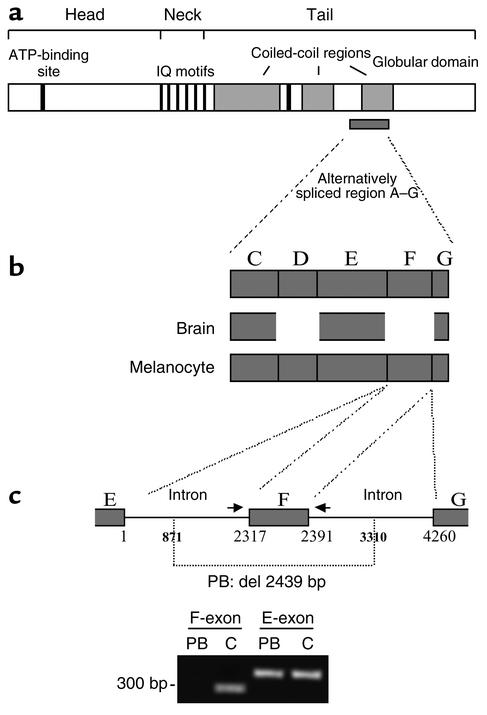
Griscelli syndrome (GS) is a rare autosomal recessive disorder that associates hypopigmentation, characterized by a silver-gray sheen of the hair and the presence of large clusters of pigment in the hair shaft, and the occurrence of either a primary neurological impairment or a severe immune disorder. Two different genetic forms, GS1 and GS2, respectively, account for the mutually exclusive neurological and immunological phenotypes. Mutations in the gene encoding the molecular motor protein Myosin Va (MyoVa) cause GS1 and the dilute mutant in mice, whereas mutations in the gene encoding the small GTPase Rab27a are responsible for GS2 and the ashen mouse model. We herein present genetic and functional evidence that a third form of GS (GS3), whose expression is restricted to the characteristic hypopigmentation of GS, results from mutation in the gene that encodes melanophilin (Mlph), the ortholog of the gene mutated in leaden mice. We also show that an identical phenotype can result from the deletion of the MYO5A F-exon, an exon with a tissue-restricted expression pattern. This spectrum of GS conditions pinpoints the distinct molecular pathways used by melanocytes, neurons, and immune cells in secretory granule exocytosis, which in part remain to be unraveled.
Figures





References
-
- Griscelli C, et al. A syndrome associating partial albinism and immunodeficiency. Am. J. Med. 1978;65:691–702. - PubMed
-
- Pastural E, et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat. Genet. 1997;16:289–292. - PubMed
-
- Ménasché G, et al. Mutations in RAB27A cause Griscelli syndrome associated with hemophagocytic syndrome. Nat. Genet. 2000;25:173–176. - PubMed
-
- Langford GM, Molyneaux BJ. Myosin V in the brain: mutations lead to neurological defects. Brain Res. Brain Res. Rev. 1998;28:1–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
