

VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization
- PMID: 12900522
- PMCID: PMC2194095
- DOI: 10.1084/jem.20022027
VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization
Abstract
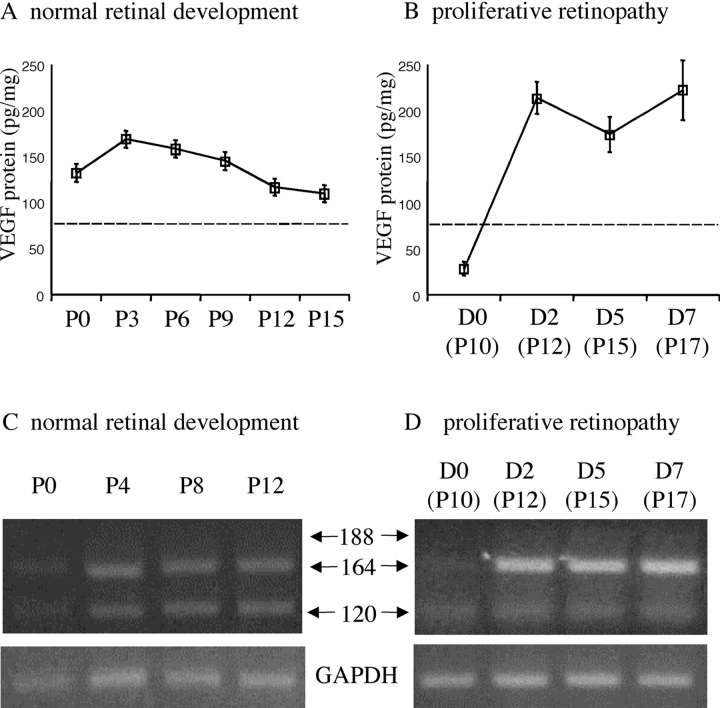
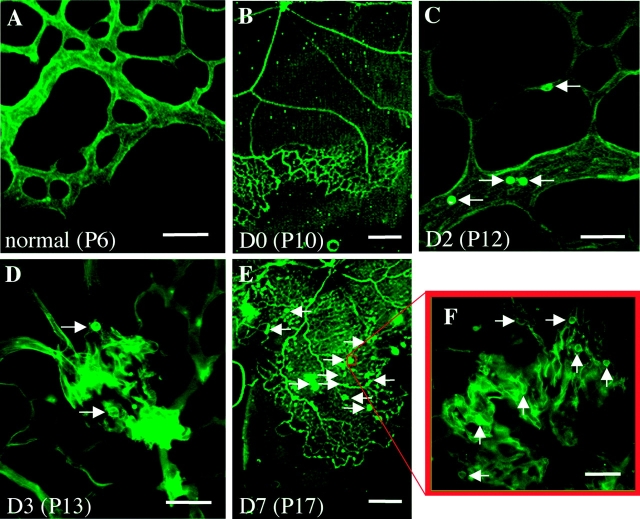
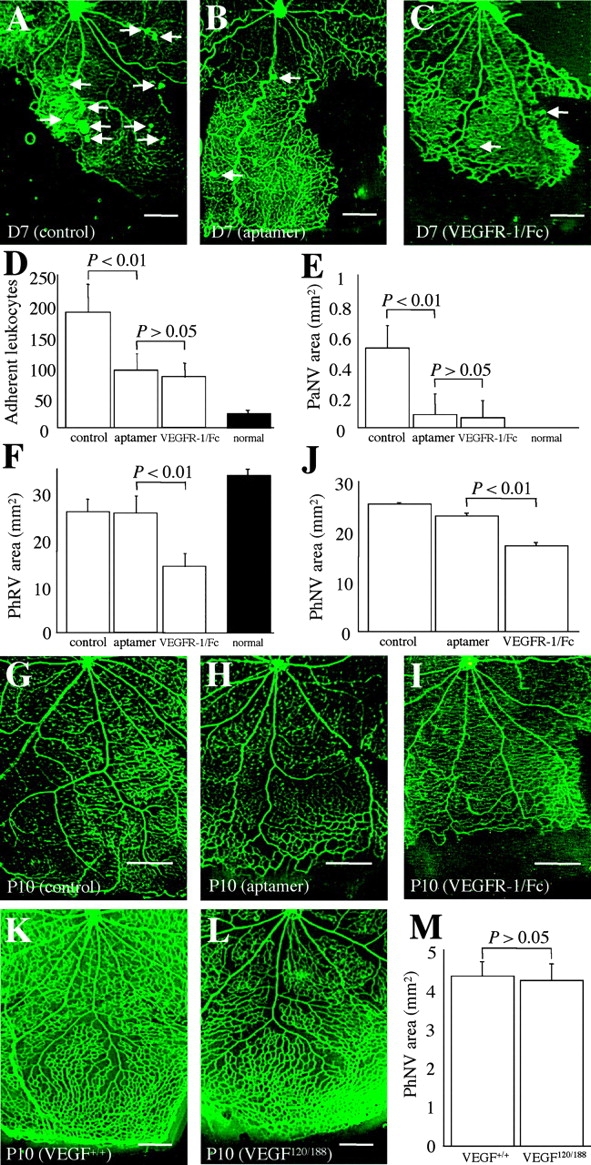
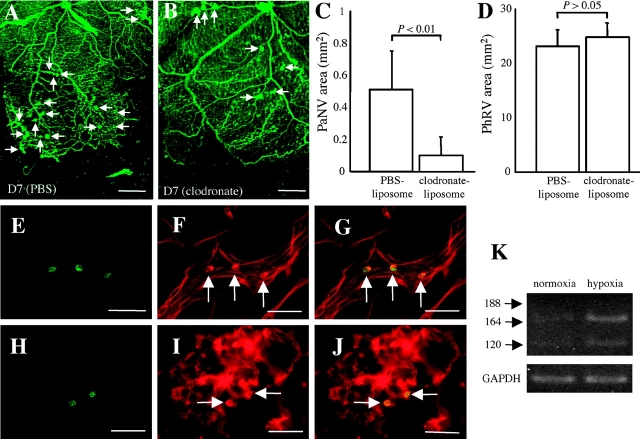
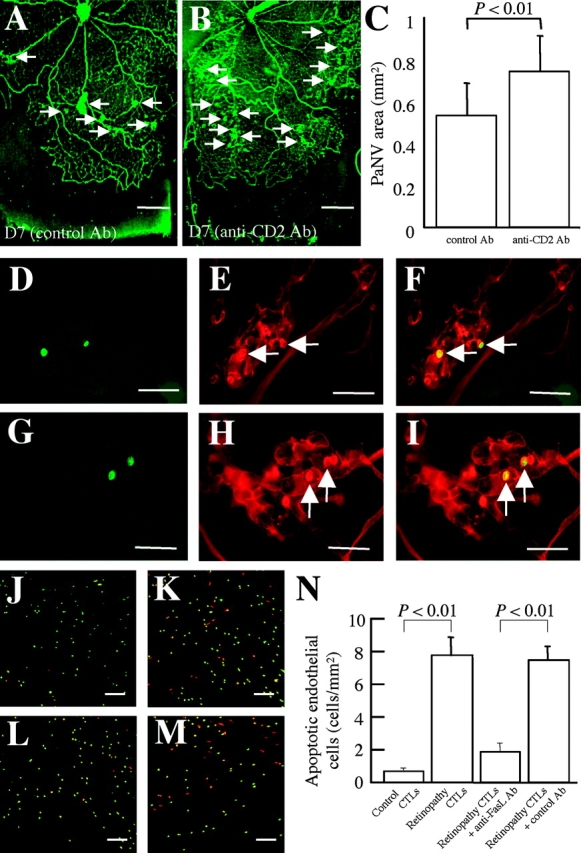
Hypoxia-induced VEGF governs both physiological retinal vascular development and pathological retinal neovascularization. In the current paper, the mechanisms of physiological and pathological neovascularization are compared and contrasted. During pathological neovascularization, both the absolute and relative expression levels for VEGF164 increased to a greater degree than during physiological neovascularization. Furthermore, extensive leukocyte adhesion was observed at the leading edge of pathological, but not physiological, neovascularization. When a VEGF164-specific neutralizing aptamer was administered, it potently suppressed the leukocyte adhesion and pathological neovascularization, whereas it had little or no effect on physiological neovascularization. In parallel experiments, genetically altered VEGF164-deficient (VEGF120/188) mice exhibited no difference in physiological neovascularization when compared with wild-type (VEGF+/+) controls. In contrast, administration of a VEGFR-1/Fc fusion protein, which blocks all VEGF isoforms, led to significant suppression of both pathological and physiological neovascularization. In addition, the targeted inactivation of monocyte lineage cells with clodronate-liposomes led to the suppression of pathological neovascularization. Conversely, the blockade of T lymphocyte-mediated immune responses with an anti-CD2 antibody exacerbated pathological neovascularization. These data highlight important molecular and cellular differences between physiological and pathological retinal neovascularization. During pathological neovascularization, VEGF164 selectively induces inflammation and cellular immunity. These processes provide positive and negative angiogenic regulation, respectively. Together, new therapeutic approaches for selectively targeting pathological, but not physiological, retinal neovascularization are outlined.
Figures
References
-
- Stone, J., T. Chan-Ling, J. Pe'er, A. Itin, H. Gnessin, and E. Keshet. 1996. Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 37:290–299. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
