

A population threshold for functional polymorphisms
- PMID: 12902381
- PMCID: PMC403778
- DOI: 10.1101/gr.1324303
A population threshold for functional polymorphisms
Abstract
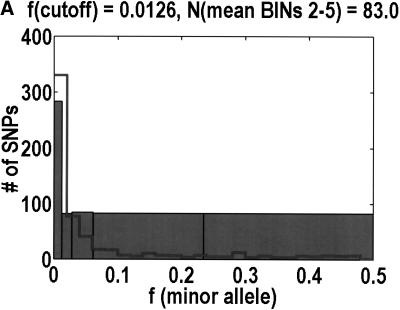
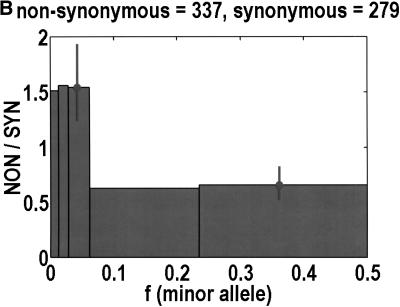
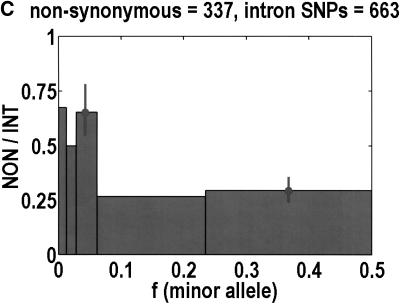
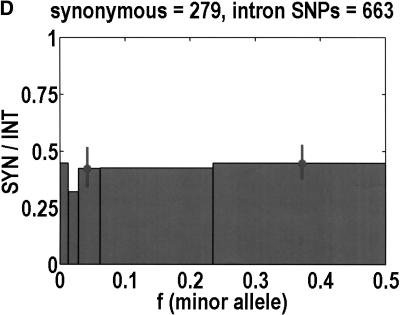
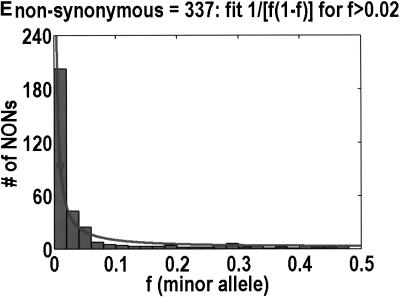
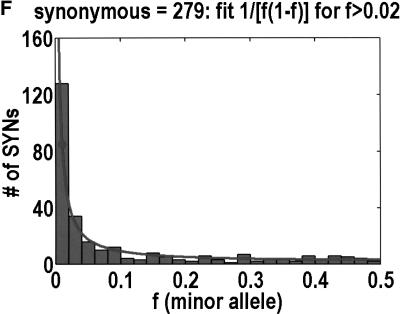
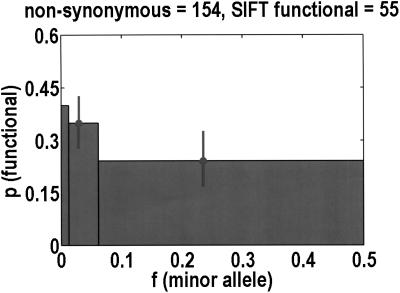
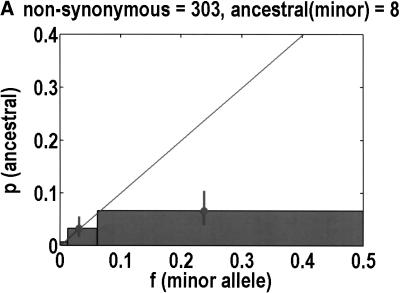
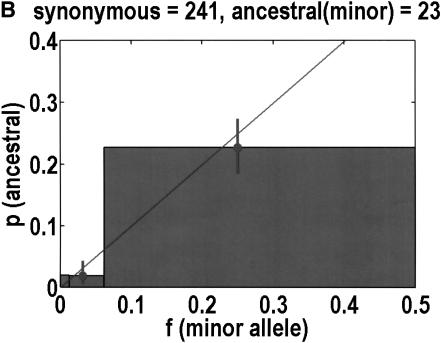
We sequenced 114 genes (for DNA repair, cell cycle arrest, apoptosis, and detoxification)in a mixed human population and observed a sudden increase in the number of functional polymorphisms below a minor allele frequency of approximately 6%. Functionality is assessed by considering the ratio in the number of nonsynonymous single nucletide polymorphisms (SNPs)to the number of synonymous or intron SNPs. This ratio is steady from below 1% in frequency-that regime traditionally associated with rare Mendelian diseases-all the way up to about 6% in frequency, after which it falls precipitously. We consider possible explanations for this threshold effect. There are four candidates as follows: (1). deleterious variants that have yet to be purified from the population, (2). balancing selection, in which a selective advantage accrues to the heterozygotes, (3). population-specific functional polymorphisms, and (4). adaptive variants that are accumulating in the population as a response to the dramatic environmental changes of the last 7000 approximately 17000 years.
Figures
References
-
- Bailey, J.A., Gu, Z., Clark, R.A., Reinert, K., Samonte, R.V., Schwartz, S., Adams, M.D., Myers, E.W., Li, P.W., and Eichler, E.E. 2002. Recent segmental duplications in the human genome. Science 297: 1003-1007. - PubMed
-
- Cargill, M., Altshuler, D., Ireland, J., Sklar, P., Ardlie, K., Patil, N., Shaw, N., Lane, C.R., Lim, E.P., Kalyanaraman, N., et al. 1999. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 22: 231-238. - PubMed
-
- Collins, F.S., Brooks, L.D., and Chakravarti, A. 1998. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 8: 1229-1231. - PubMed
WEB SITE REFERENCES
-
- http://www.genome.washington.edu/projects/egpsnps; University of Washington Genome Center Repository of Candidate-Gene Polymorphisms for Environmental Genome Project (EGP).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources