

Tumor necrosis factor-alpha at the crossroads of neuronal life and death during HIV-associated dementia
- PMID: 12911614
- PMCID: PMC1955474
- DOI: 10.1046/j.1471-4159.2003.01942.x
Tumor necrosis factor-alpha at the crossroads of neuronal life and death during HIV-associated dementia
Abstract
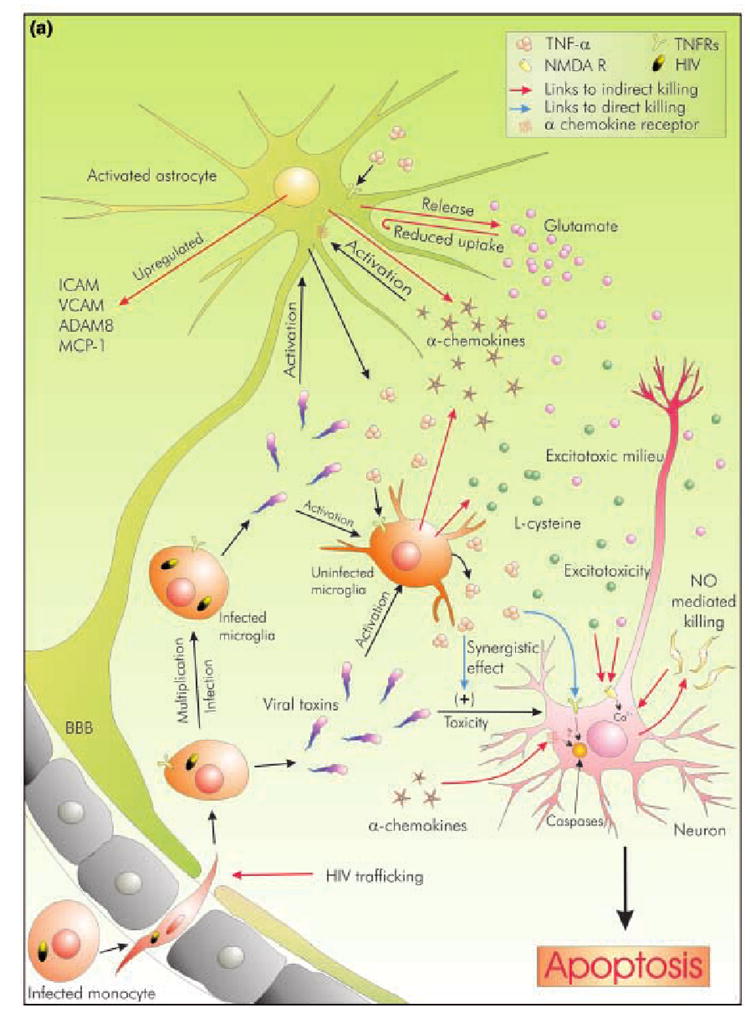
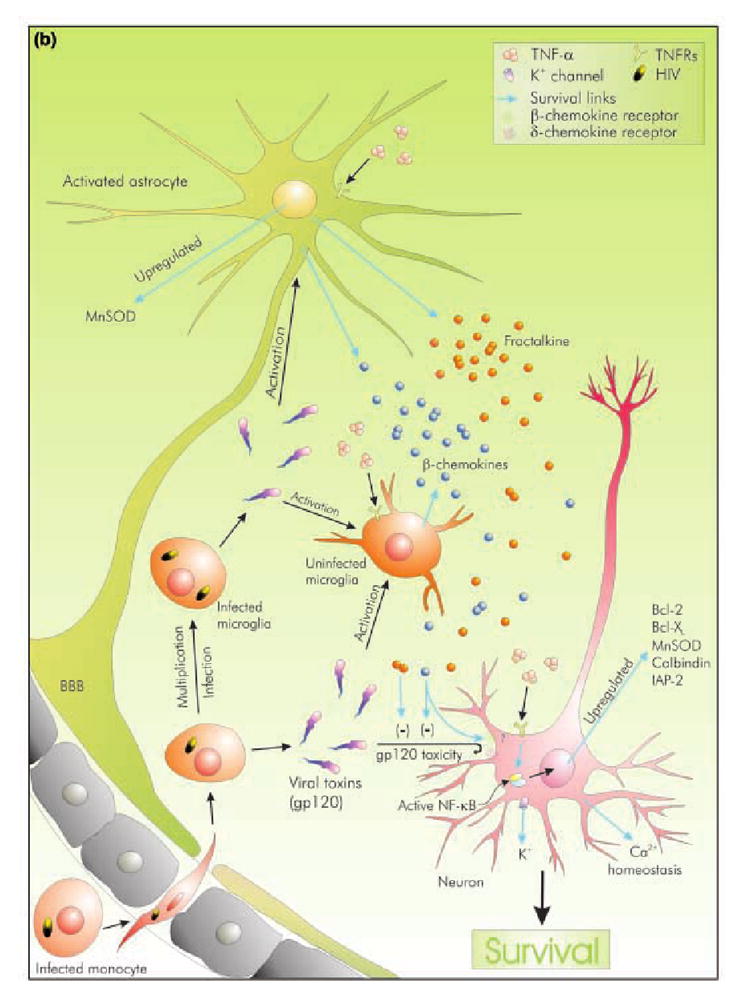
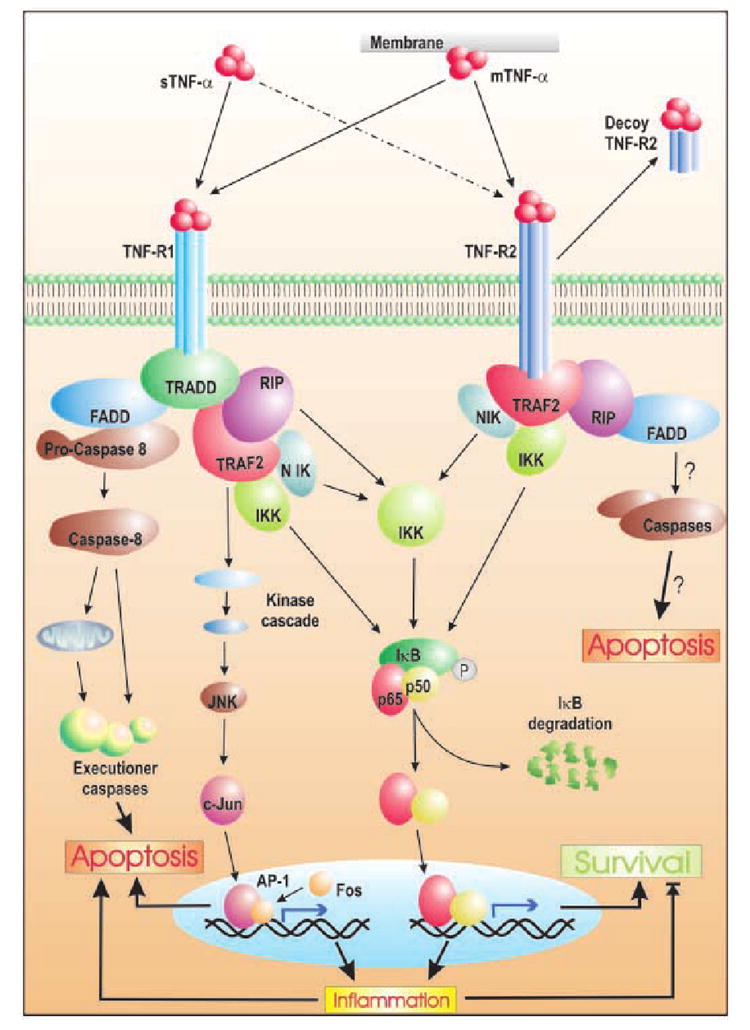
Human immunodeficiency type-1 (HIV-1) infection is known to cause disorders of the CNS, including HIV-associated dementia (HAD). It is suspected that tumor necrosis factor-alpha (TNF-alpha) released by infected microglia and macrophages play a role in neuronal injury seen in HAD patients. Accordingly, studies suggest that the level of TNF-alpha mRNA increases with increasing severity of dementia in patients, and that inhibitors of TNF-alpha release reduces neuronal injury in murine model of HAD. However, the exact role of TNF-alpha in relation to neuronal dysfunction is a matter of ongoing debate. One school of thought hails TNF-alpha as the inducer and mediator of neurodegeneration and their evidence suggest that TNF-alpha kill neurons directly by recruiting caspases or may kill indirectly by various means. In sharp contrast to this, another concept theory envisages a role for TNF-alpha in negotiating neuroprotection during HAD. The current compilation examines these contradictory concepts, and evaluates their efficacy in the light of TNF-alpha signaling. It also attempts to elaborate the current consensus outlook of TNF-alpha's role during HAD.
Figures
Similar articles
-
The signaling and apoptotic effects of TNF-related apoptosis-inducing ligand in HIV-1 associated dementia.Neurotox Res. 2005 Oct;8(1-2):135-48. doi: 10.1007/BF03033825. Neurotox Res. 2005. PMID: 16260391 Review.
-
Human immunodeficiency virus type-1 protein Tat induces tumor necrosis factor-alpha-mediated neurotoxicity.Neurobiol Dis. 2007 Jun;26(3):661-70. doi: 10.1016/j.nbd.2007.03.004. Epub 2007 Mar 20. Neurobiol Dis. 2007. PMID: 17451964 Free PMC article.
-
Similarity of neuronal cell injury and death in AIDS dementia and focal cerebral ischemia: potential treatment with NMDA open-channel blockers and nitric oxide-related species.Brain Pathol. 1996 Oct;6(4):507-17. doi: 10.1111/j.1750-3639.1996.tb00879.x. Brain Pathol. 1996. PMID: 8944320 Review.
-
Neuronal PINCH is regulated by TNF-α and is required for neurite extension.J Neuroimmune Pharmacol. 2011 Sep;6(3):330-40. doi: 10.1007/s11481-010-9236-5. Epub 2010 Aug 6. J Neuroimmune Pharmacol. 2011. PMID: 20689998 Free PMC article.
-
In vitro evidence for a dual role of tumor necrosis factor-alpha in human immunodeficiency virus type 1 encephalopathy.Ann Neurol. 1995 Mar;37(3):381-94. doi: 10.1002/ana.410370315. Ann Neurol. 1995. PMID: 7695238
Cited by
-
Ibudilast, a pharmacologic phosphodiesterase inhibitor, prevents human immunodeficiency virus-1 Tat-mediated activation of microglial cells.PLoS One. 2011 Apr 8;6(4):e18633. doi: 10.1371/journal.pone.0018633. PLoS One. 2011. PMID: 21494611 Free PMC article.
-
Silver Citrate Nanoparticles Inhibit PMA-Induced TNFα Expression via Deactivation of NF-κB Activity in Human Cancer Cell-Lines, MCF-7.Int J Nanomedicine. 2020 Oct 30;15:8479-8493. doi: 10.2147/IJN.S274098. eCollection 2020. Int J Nanomedicine. 2020. PMID: 33154638 Free PMC article.
-
Proinflammatory cytokines and HIV-1 synergistically enhance CXCL10 expression in human astrocytes.Glia. 2009 May;57(7):734-43. doi: 10.1002/glia.20801. Glia. 2009. PMID: 18985732 Free PMC article.
-
Inflammatory mediators reduce surface PrPc on human BMVEC resulting in decreased barrier integrity.Lab Invest. 2018 Oct;98(10):1347-1359. doi: 10.1038/s41374-018-0090-z. Epub 2018 Jun 29. Lab Invest. 2018. PMID: 29959417 Free PMC article.
-
HIV-associated synaptic degeneration.Mol Brain. 2017 Aug 29;10(1):40. doi: 10.1186/s13041-017-0321-z. Mol Brain. 2017. PMID: 28851400 Free PMC article. Review.
References
-
- Adle-Biassette H, Levy Y, Colombel M, Poron F, Natchev S, Keohane C, Gray F. Neuronal apoptosis in HIV infection in adults. Neurpathol Appl Neurobiol. 1995;21:218–227. - PubMed
-
- Adle-Biassette H, Chretien F, Wingertsmann L, Hery C, Ereau T, Scaravilli F, Tardieu M, Gray F. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25:123–133. - PubMed
-
- Andjelkovic AV, Kerkovich D, Shanley J, Pulliam L, Pachter JS. Expression of binding sites for β chemokines on human astrocytes. Glia. 1999;28:225–235. - PubMed
-
- Asensio VC, Campbell IL. Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci. 1999;22:504–512. - PubMed
-
- Bal-Price A, Moneer Z, Brown GC. Nitric oxide induces rapid, calcium dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia. 2002;40:312–323. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
