

Reversal of human cellular senescence: roles of the p53 and p16 pathways
- PMID: 12912919
- PMCID: PMC175806
- DOI: 10.1093/emboj/cdg417
Reversal of human cellular senescence: roles of the p53 and p16 pathways
Abstract
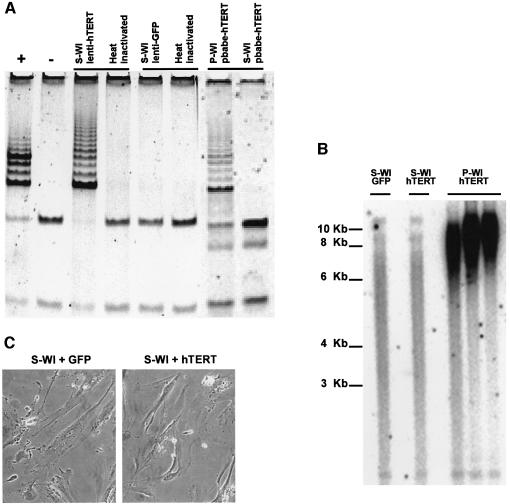
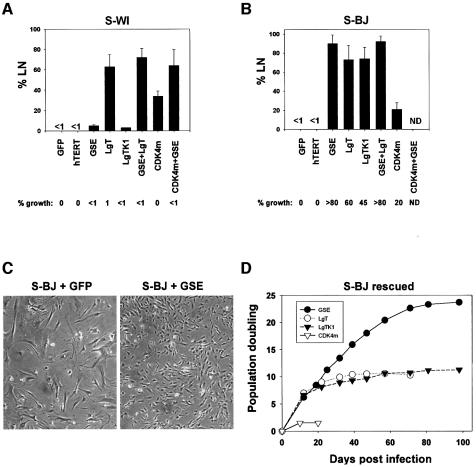
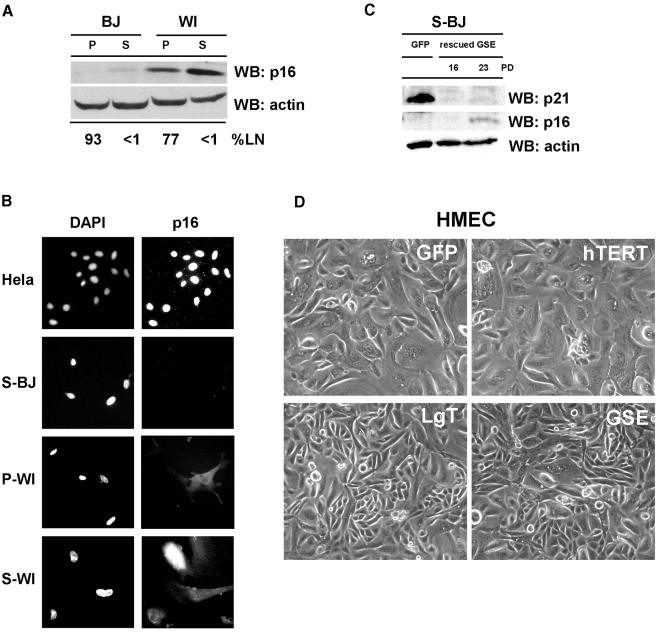
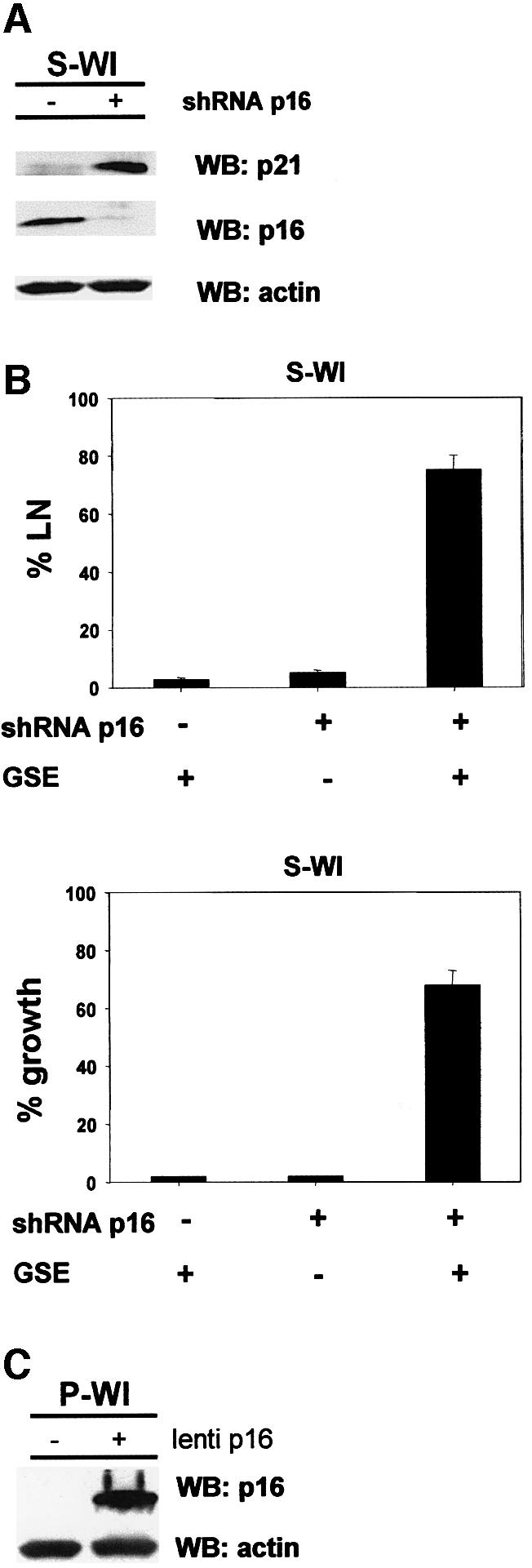
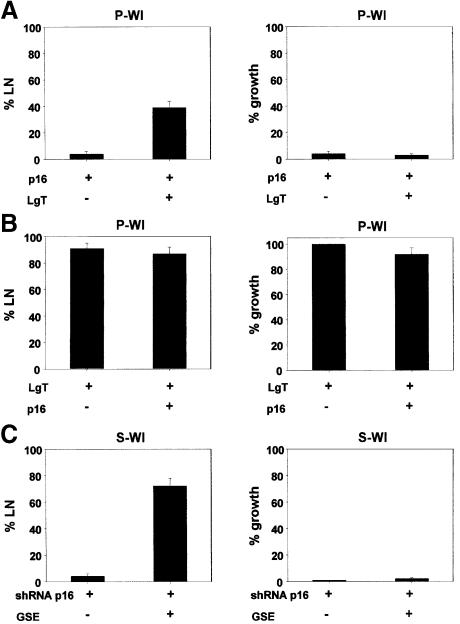
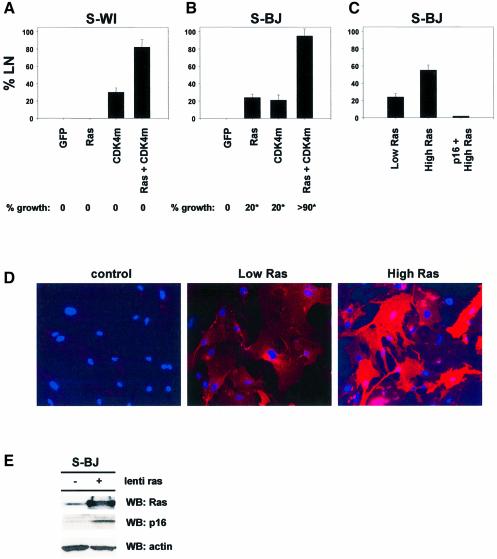
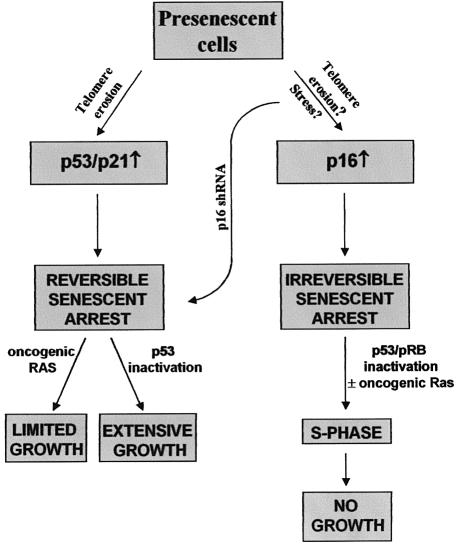
Telomere erosion and subsequent dysfunction limits the proliferation of normal human cells by a process termed replicative senescence. Replicative senescence is thought to suppress tumorigenesis by establishing an essentially irreversible growth arrest that requires activities of the p53 and pRB tumor suppressor proteins. We show that, depending on expression of the pRB regulator p16, replicative senescence is not necessarily irreversible. We used lentiviruses to express specific viral and cellular proteins in senescent human fibroblasts and mammary epithelial cells. Expression of telomerase did not reverse the senescence arrest. However, cells with low levels of p16 at senescence resumed robust growth upon p53 inactivation, and limited growth upon expression of oncogenic RAS. In contrast, cells with high levels of p16 at senescence failed to proliferate upon p53 inactivation or RAS expression, although they re-entered the cell cycle without growth after pRB inactivation. Our results indicate that the senescence response to telomere dysfunction is reversible and is maintained primarily by p53. However, p16 provides a dominant second barrier to the unlimited growth of human cells.
Figures
References
-
- Blackburn E.H. (2001) Switching and signaling at the telomere. Cell, 106, 661–673. - PubMed
-
- Bodnar A.G. et al. (1998) Extension of life span by introduction of telomerase into normal human cells. Science, 279, 349–352. - PubMed
-
- Brehm A. and Kouzarides,T. (1999) Retinoblastoma protein meets chromatin. Trends Biochem. Sci., 24, 142–145. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
