

Link between genome packaging and rate of budding for Rous sarcoma virus
- PMID: 12915554
- PMCID: PMC187400
- DOI: 10.1128/jvi.77.17.9388-9398.2003
Link between genome packaging and rate of budding for Rous sarcoma virus
Abstract
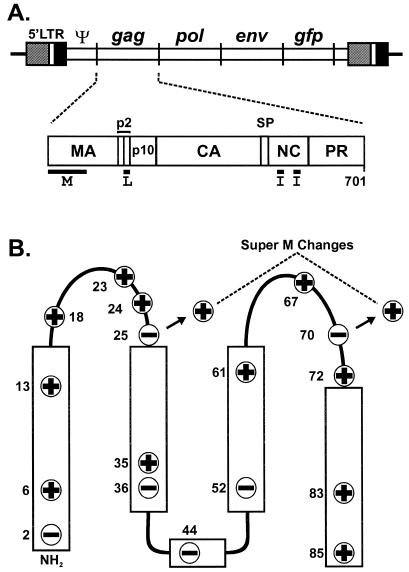
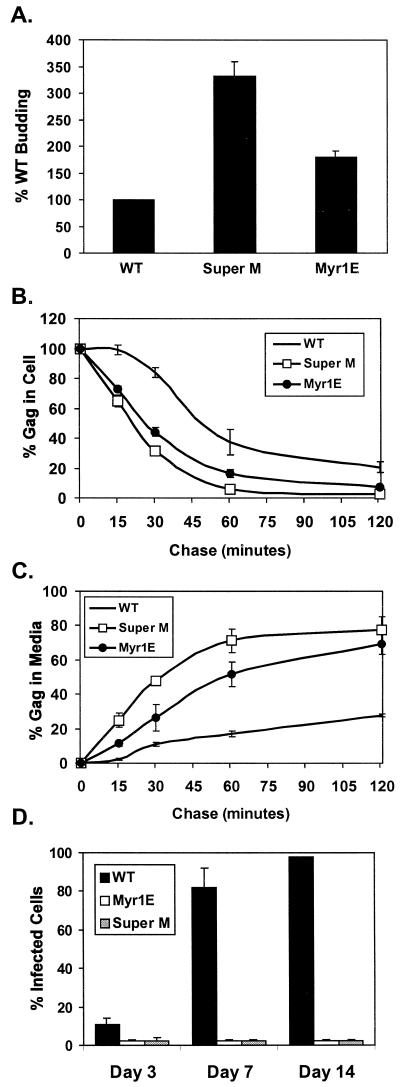
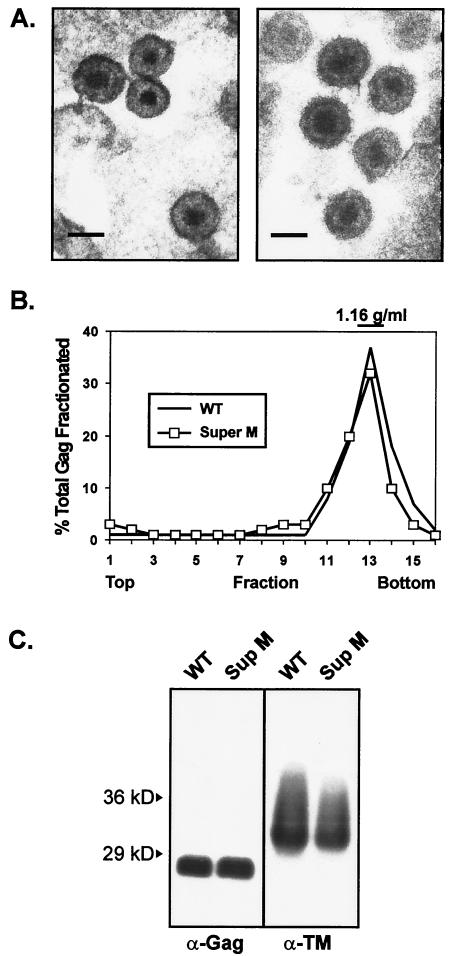
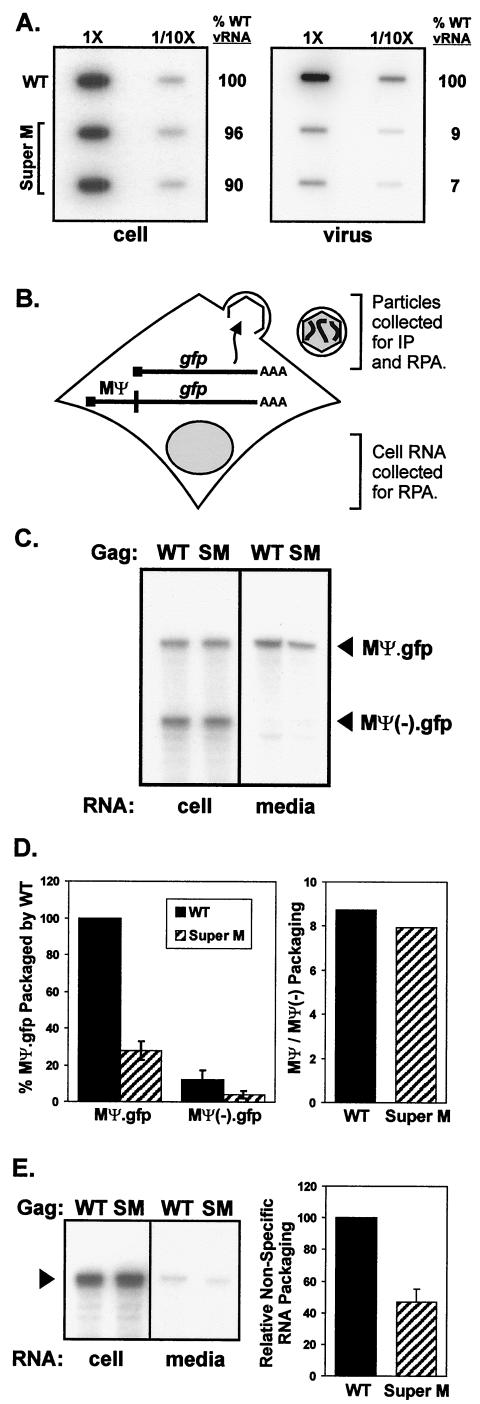
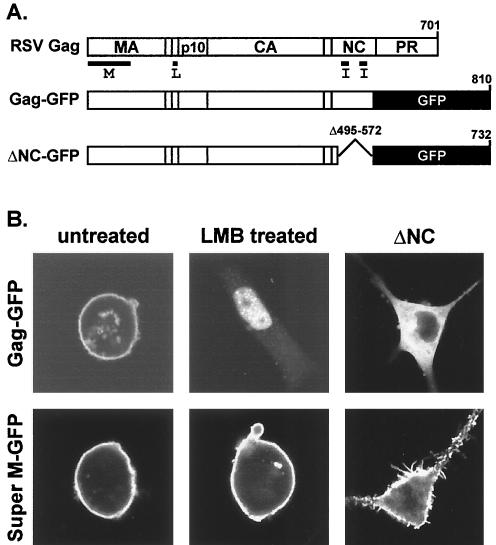
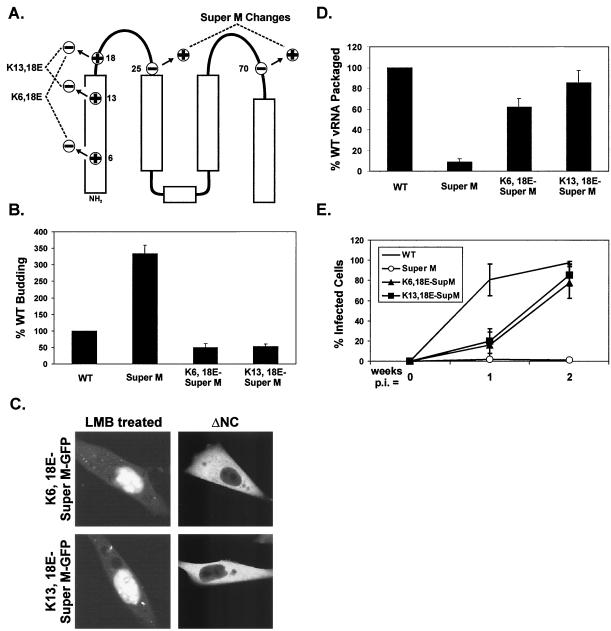
The subcellular location at which genomic RNA is packaged by Gag proteins during retrovirus assembly remains unknown. Since the membrane-binding (M) domain is most critical for targeting Gag to the plasma membrane, changes to this determinant might alter the path taken through the cell and reduce the efficiency of genome packaging. In this report, a Rous sarcoma virus (RSV) mutant having two acidic-to-basic substitutions in the M domain is described. This mutant, designated Super M, produced particles much faster than the wild type, but the mutant virions were noninfectious and contained only 1/10 the amount of genomic RNA found in wild-type particles. To identify the cause(s) of these defects, we considered data that suggest that RSV Gag traffics through the nucleus to package the viral genome. Although inhibition of the CRM-1 pathway of nuclear export caused the accumulation of wild-type Gag in the nucleus, nuclear accumulation did not occur with Super M. The importance of the nucleocapsid (NC) domain in membrane targeting was also determined, and, importantly, deletion of the NC sequence prevented plasma membrane localization by wild-type Gag but not by Super M Gag. Based on these results, we reasoned that the enhanced membrane-targeting properties of Super M inhibit genome packaging. Consistent with this interpretation, substitutions that reestablished the wild-type number of basic and acidic residues in the Super M Gag M domain reduced the budding efficiency and restored genome packaging and infectivity. Therefore, these data suggest that Gag targeting and genome packaging are normally linked to ensure that RSV particles contain viral RNA.
Figures
References
-
- Burniston, M. T., A. Cimarelli, J. Colgan, S. P. Curtis, and J. Luban. 1999. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J. Virol. 73:8527-8540. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
