

Structural and phylogenetic analysis of adenovirus hexons by use of high-resolution x-ray crystallographic, molecular modeling, and sequence-based methods
- PMID: 12915569
- PMCID: PMC187380
- DOI: 10.1128/jvi.77.17.9553-9566.2003
Structural and phylogenetic analysis of adenovirus hexons by use of high-resolution x-ray crystallographic, molecular modeling, and sequence-based methods
Abstract
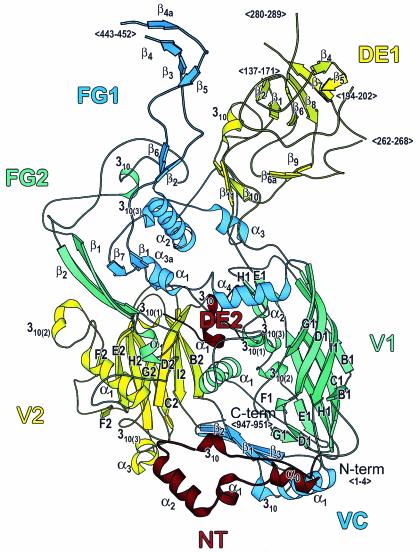
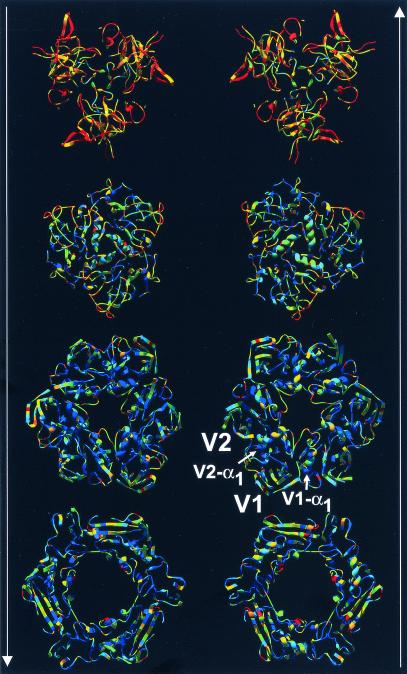
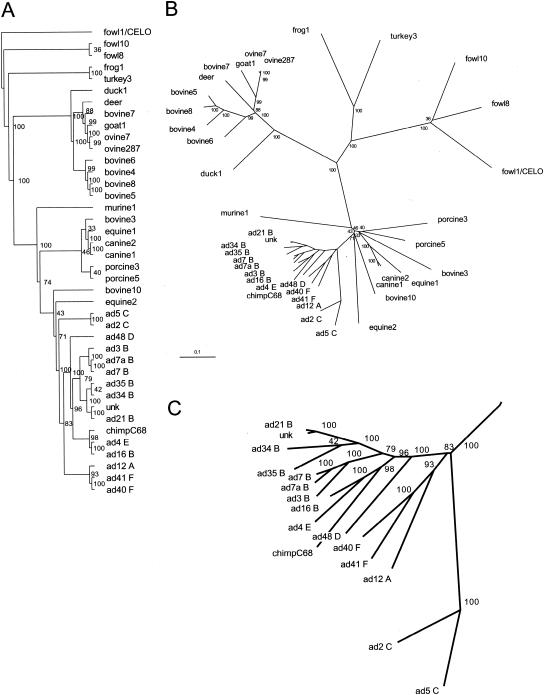
A major impediment to the use of adenovirus as a gene therapy vector and for vaccine applications is the host immune response to adenovirus hexon-the major protein component of the icosahedral capsid. A solution may lie in novel vectors with modified or chimeric hexons designed to evade the immune response. To facilitate this approach, we have distinguished the portion of hexon that all serotypes have in common from the hypervariable regions that are responsible for capsid diversity and type-specific immunogenicity. The common hexon core-conserved because it forms the viral capsid-sets boundaries to the regions where modifications can be made to produce nonnative hexons. The core has been defined from the large and diverse set of known hexon sequences by an accurate alignment based on the newly refined crystal structures of human adenovirus types 2 (Ad2) and Ad5 hexon. Comparison of the two hexon models, which are the most accurate so far, reveals that over 90% of the residues in each have three-dimensional positions that closely match. Structures for more distant hexons were predicted by building molecular models of human Ad4, chimpanzee adenovirus (AdC68), and fowl adenovirus 1 (FAV1 or CELO). The five structures were then used to guide the alignment of the 40 full-length (>900 residues) hexon sequences in public databases. Distance- and parsimony-based phylogenetic trees are consistent and reveal evolutionary relationships between adenovirus types that parallel those of their animal hosts. The combination of crystallography, molecular modeling, and phylogenetic analysis defines a conserved molecular core that can serve as the armature for the directed design of novel hexons.
Figures


References
-
- Athappilly, F. K., R. Murali, J. J. Rux, Z. Cai, and R. M. Burnett. 1994. The refined crystal structure of hexon, the major coat protein of adenovirus type 2, at 2.9 Å resolution. J. Mol. Biol. 242:430-455. - PubMed
-
- Bamford, D. H., R. M. Burnett, and D. I. Stuart. 2002. Evolution of viral structure. Theor. Pop. Biol. 61:461-470. - PubMed
-
- Benkö, M., B. Harrach, and J. C. D'Halluin. 1990. Molecular cloning and physical mapping of the DNA of bovine adenovirus serotype 4; study of the DNA homology among bovine, human, and porcine adenoviruses. J. Gen. Virol. 71:465-469. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
