

Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections
- PMID: 12925671
- PMCID: PMC2194174
- DOI: 10.1084/jem.20030701
Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections
Abstract
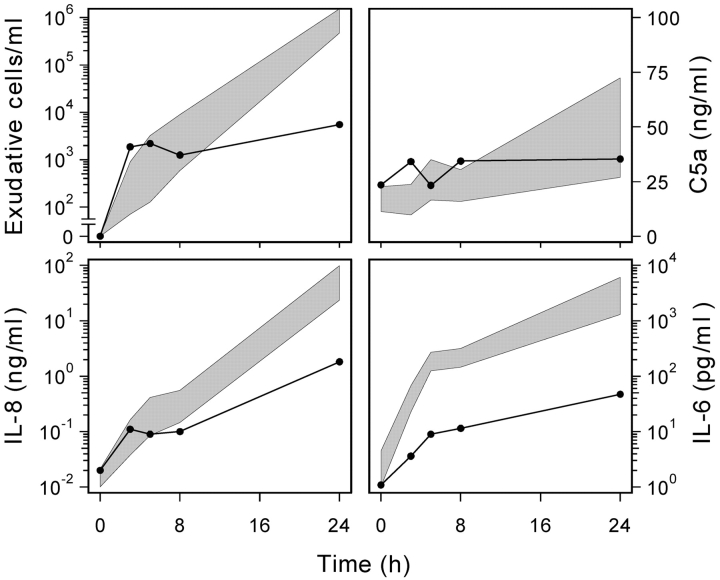
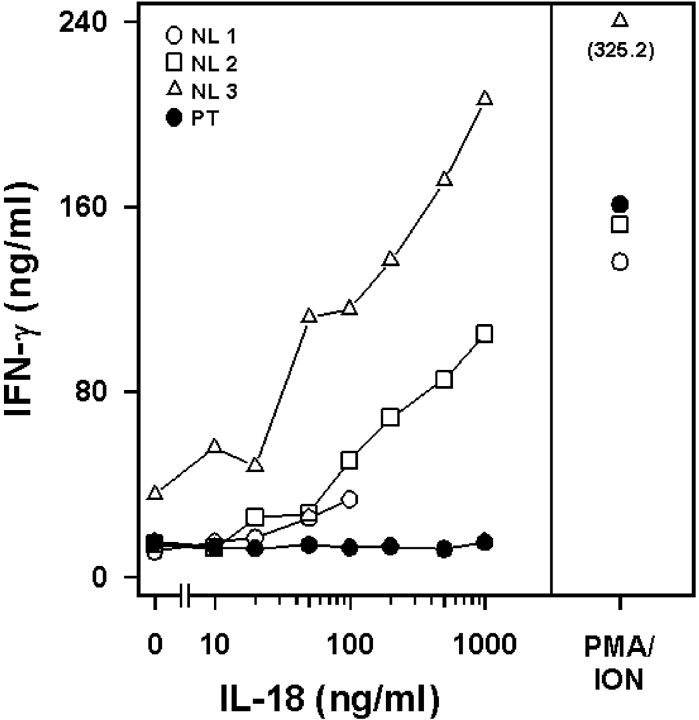
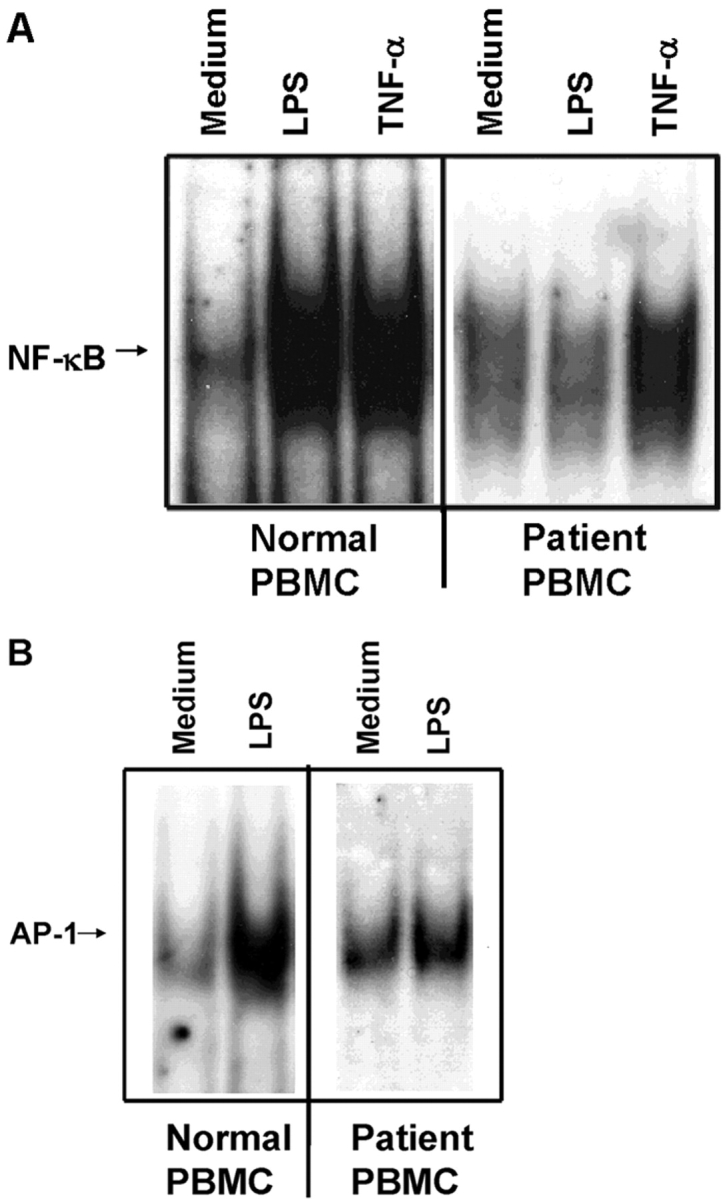
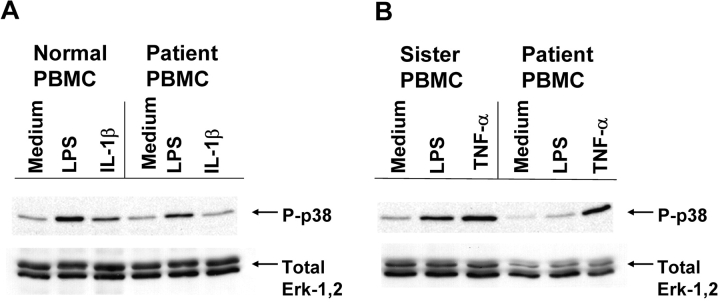
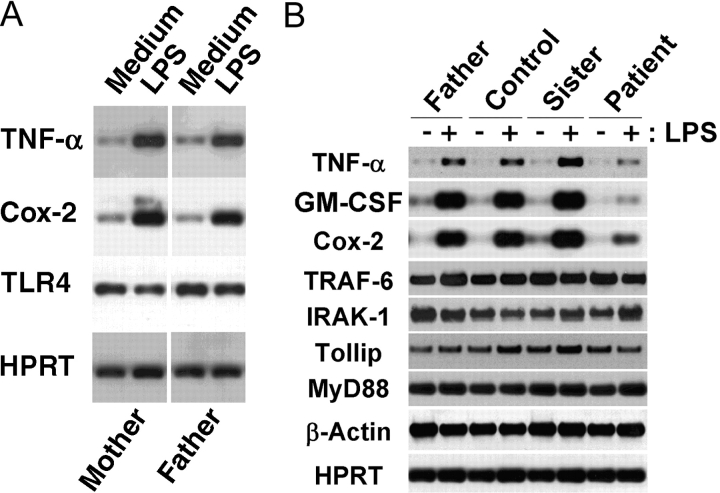
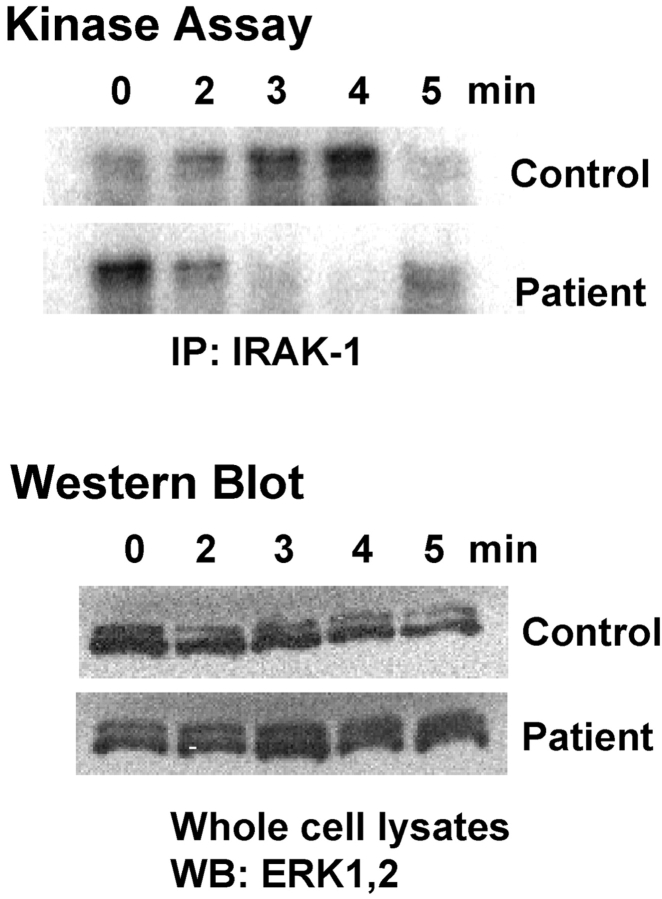
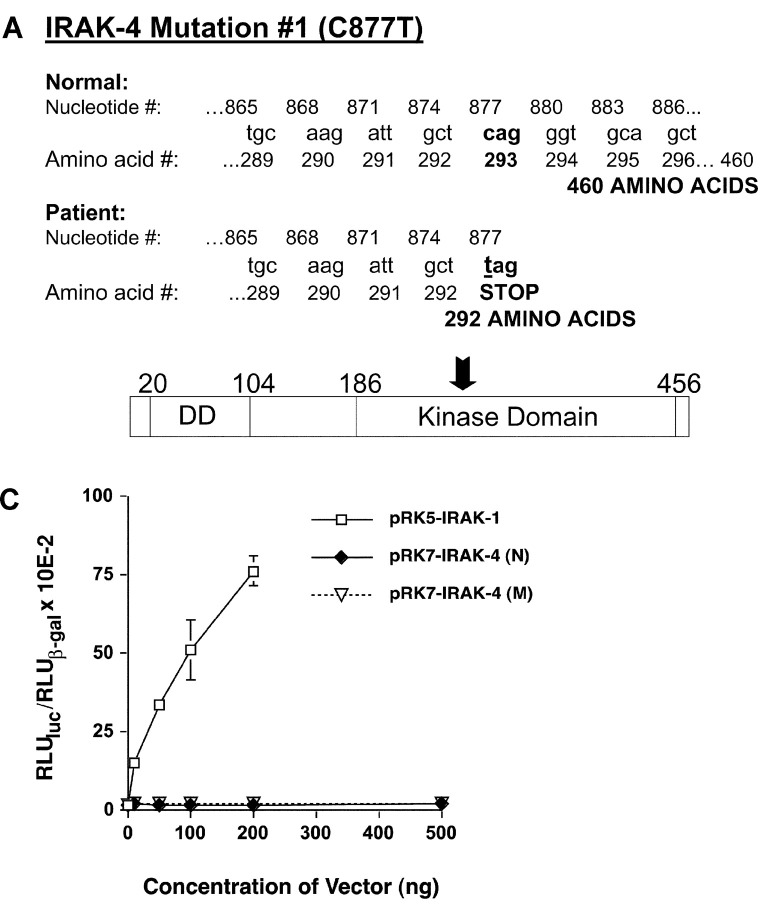
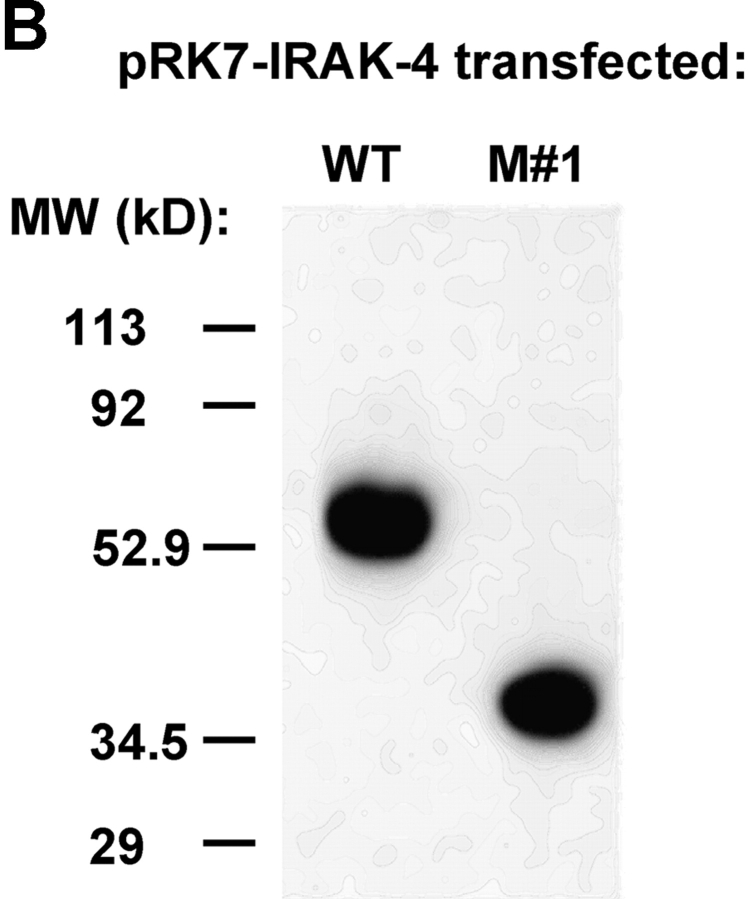
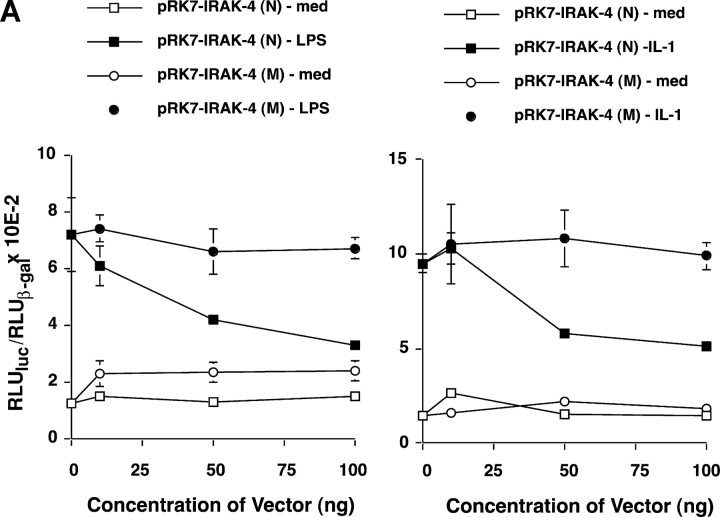
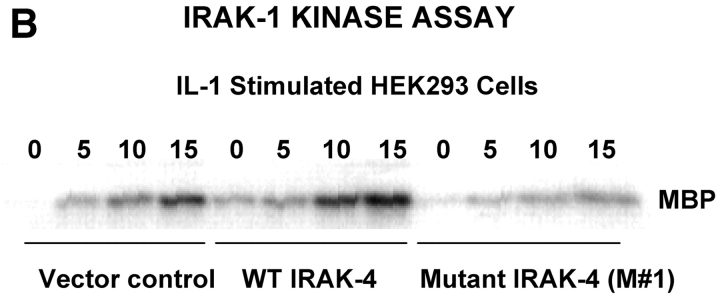
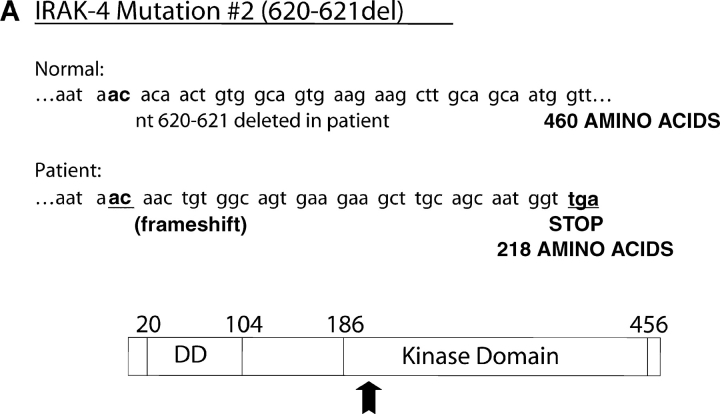
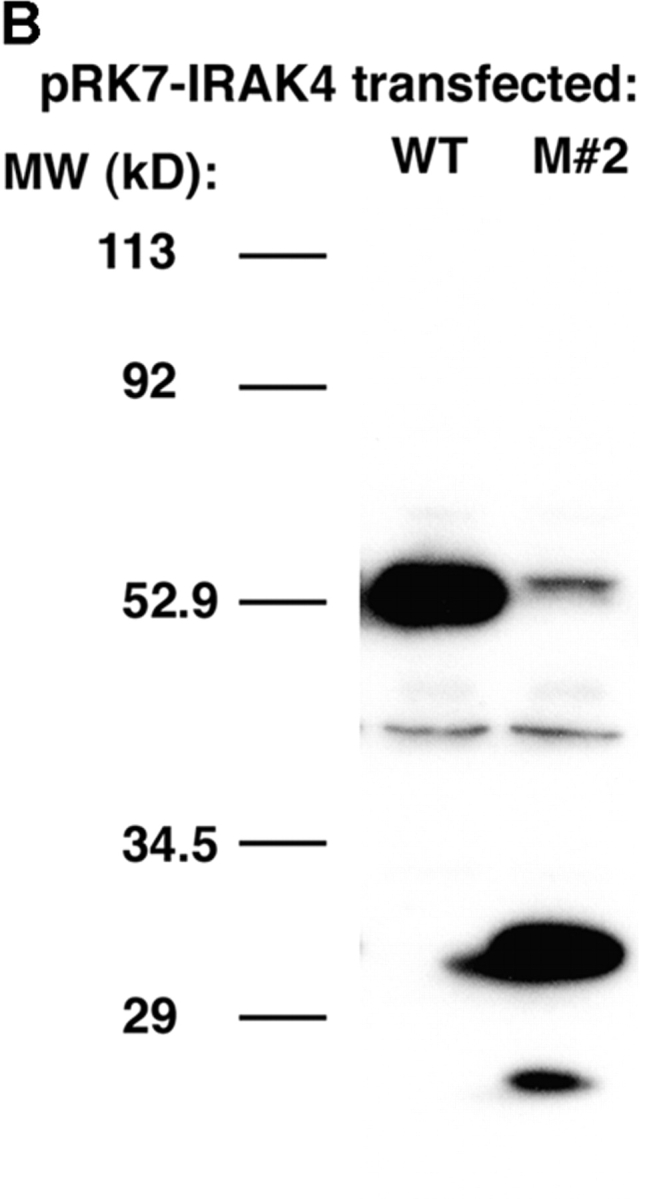
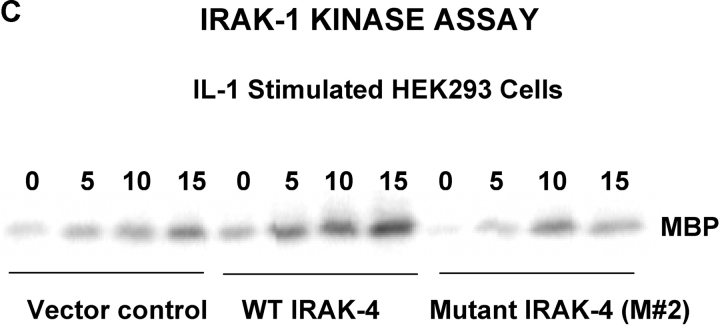
We identified previously a patient with recurrent bacterial infections who failed to respond to gram-negative LPS in vivo, and whose leukocytes were profoundly hyporesponsive to LPS and IL-1 in vitro. We now demonstrate that this patient also exhibits deficient responses in a skin blister model of aseptic inflammation. A lack of IL-18 responsiveness, coupled with diminished LPS and/or IL-1-induced nuclear factor-kappaB and activator protein-1 translocation, p38 phosphorylation, gene expression, and dysregulated IL-1R-associated kinase (IRAK)-1 activity in vitro support the hypothesis that the defect lies within the signaling pathway common to toll-like receptor 4, IL-1R, and IL-18R. This patient expresses a "compound heterozygous" genotype, with a point mutation (C877T in cDNA) and a two-nucleotide, AC deletion (620-621del in cDNA) encoded by distinct alleles of the IRAK-4 gene (GenBank/EMBL/DDBJ accession nos. AF445802 and AY186092). Both mutations encode proteins with an intact death domain, but a truncated kinase domain, thereby precluding expression of full-length IRAK-4 (i.e., a recessive phenotype). When overexpressed in HEK293T cells, neither truncated form augmented endogenous IRAK-1 kinase activity, and both inhibited endogenous IRAK-1 activity modestly. Thus, IRAK-4 is pivotal in the development of a normal inflammatory response initiated by bacterial or nonbacterial insults.
Figures
References
-
- Kuhns, D.B., D.A. Long-Priel, and J.I. Gallin. 1997. Endotoxin and IL-1 hyporesponsiveness in a patient with recurrent bacterial infections. J. Immunol. 158:3959–3964. - PubMed
-
- Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–2088. - PubMed
-
- Medzhitov, R., P. Preston-Hurlburt, E. Kopp, A. Stadleln, C. Chen, S. Ghosh, and C.A. Janeway, Jr. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell. 2:253–258. - PubMed
-
- Medzhitov, R.P., and C.A. Janeway, Jr. 2000. Innate immunity. N. Engl. J. Med. 343:338–344. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
