

Increased mortality and dysregulated cytokine production in tumor necrosis factor receptor 1-deficient mice following systemic Klebsiella pneumoniae infection
- PMID: 12933830
- PMCID: PMC187315
- DOI: 10.1128/IAI.71.9.4891-4900.2003
Increased mortality and dysregulated cytokine production in tumor necrosis factor receptor 1-deficient mice following systemic Klebsiella pneumoniae infection
Abstract
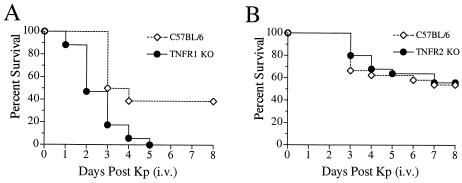
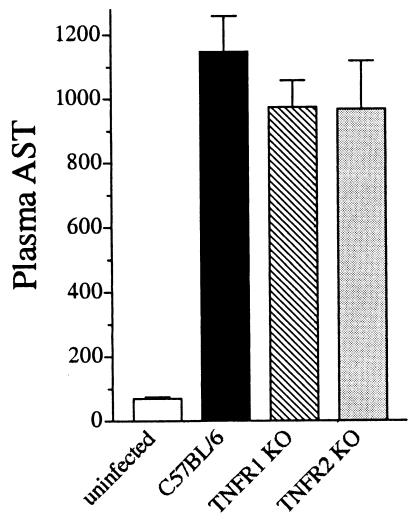
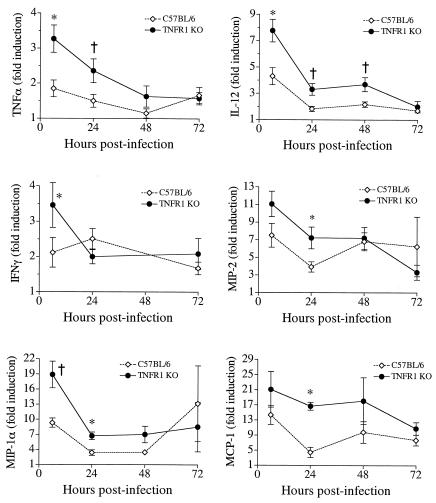
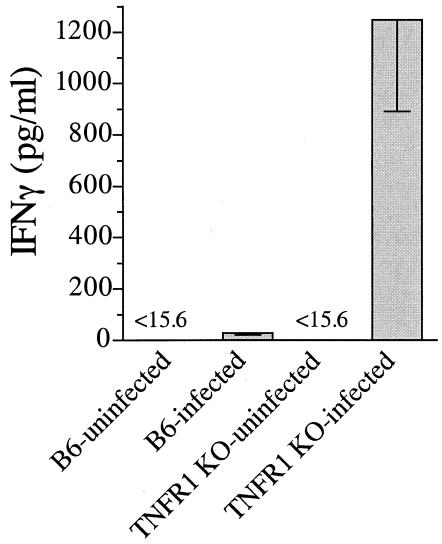
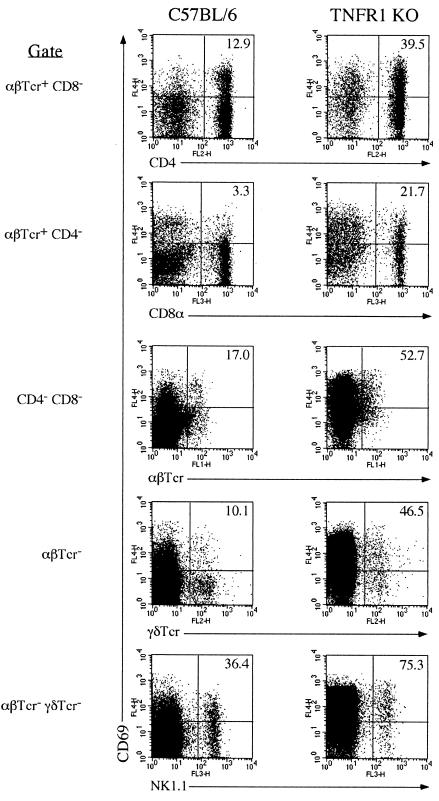
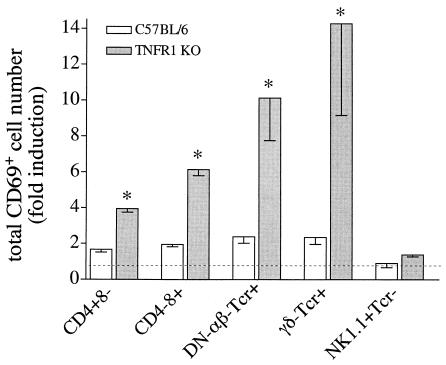
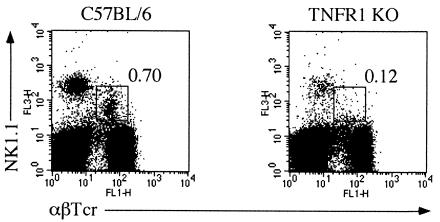
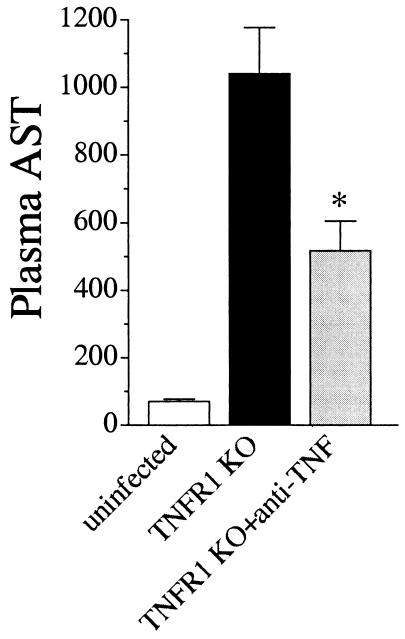
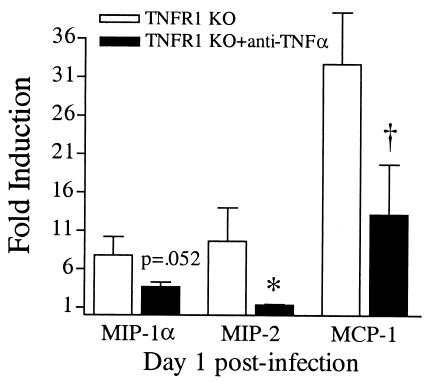
A significant clinical complication of pulmonary infections with Klebsiella pneumoniae is peripheral blood dissemination, resulting in a systemic infection concurrent with the localized pulmonary infection. In this context, little is known about the role of tumor necrosis factor receptor 1 (TNFR1)-mediated innate immune responses during systemic Klebsiella infections. Mice lacking TNFR1 were significantly more susceptible to Klebsiella-induced mortality following intravenous inoculation. Bacterial clearance was impaired in TNFR1-deficient mice at early times following infection. Unexpectedly, bacterial burdens at the onset of mortality (days 2 to 3 postinfection) were not higher in mice lacking TNFR1. However, elevated production of liver-associated proinflammatory cytokines (interleukin-12, tumor necrosis factor alpha [TNF-alpha[, and gamma interferon [IFN-gamma]) and chemokines (MIP-1 alpha, MIP-2, and MCP-1) was observed within the first 24 h of infection. Additionally, excessive plasma-associated IFN-gamma was also observed late in the course of infection (day 3). Spleen cells from day-3 infected TNFR1-deficient mice secreted markedly enhanced levels of IFN-gamma when cultured in vitro. Additionally, there was a marked increase in the total number of activated lymphocyte subsets as indicated by CD69 upregulation. A notable exception was the sharp decrease in the frequency of splenic NK T cells in infected TNFR1 knockout (KO) mice. Anti-TNF-alpha therapy in TNFR1 KO mice significantly reduced chemokine production and liver injury. Combined, these data indicate a dysregulated antibacterial host response following intravenous Klebsiella infection in the absence of TNFR1 signaling, resulting in heightened cytokine production and hyperactivation of specific splenic lymphocyte subsets.
Figures
References
-
- Allendoerfer, R., and G. S. Deepe, Jr. 2000. Regulation of infection with Histoplasma capsulatum by TNFR1 and -2. J. Immunol. 165:2657-2664. - PubMed
-
- Bank, U., and S. Ansorge. 2001. More than destructive: neutrophil-derived serine proteases in cytokine bioactivity control. J. Leukoc. Biol. 69:197-206. - PubMed
-
- Burwen, D. R., S. N. Banerjee, and R. P. Gaynes. 1994. Ceftazidime resistance among selected nosocomial gram-negative bacilli in the United States. National Nosocomial Infections Surveillance System. J. Infect. Dis. 170:1622-1625. - PubMed
-
- Carnaud, C., D. Lee, O. Donnars, S. H. Park, A. Beavis, Y. Koezuka, and A. Bendelac. 1999. Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 163:4647-4650. - PubMed
-
- Chosay, J. G., N. A. Essani, C. J. Dunn, and H. Jaeschke. 1997. Neutrophil margination and extravasation in sinusoids and venules of liver during endotoxin-induced injury. Am. J. Physiol. 272:G1195-G1200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
