

Expression deconvolution: a reinterpretation of DNA microarray data reveals dynamic changes in cell populations
- PMID: 12934019
- PMCID: PMC193568
- DOI: 10.1073/pnas.1832361100
Expression deconvolution: a reinterpretation of DNA microarray data reveals dynamic changes in cell populations
Abstract
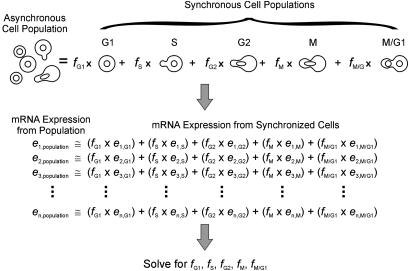
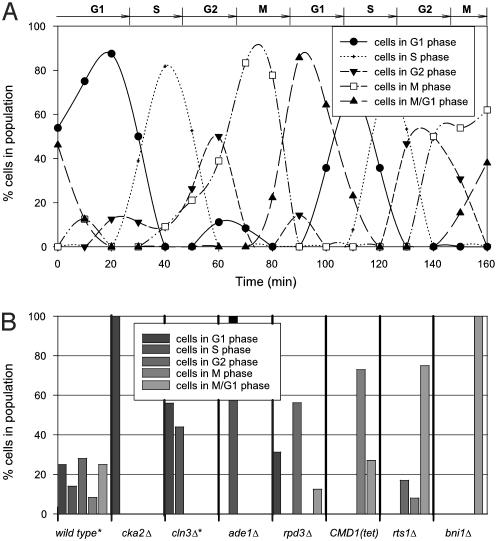
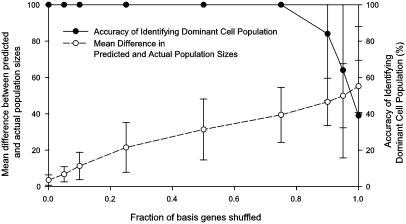
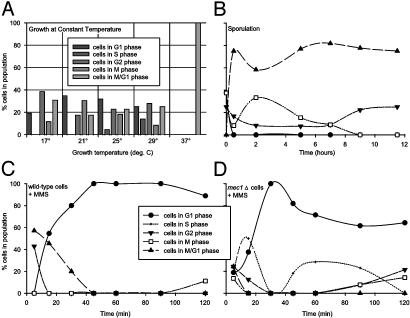
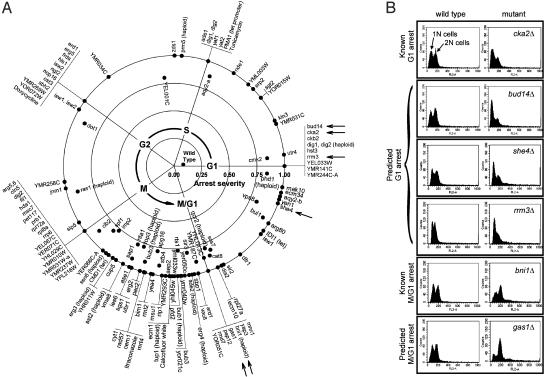
Cells grow in dynamically evolving populations, yet this aspect of experiments often goes unmeasured. A method is proposed for measuring the population dynamics of cells on the basis of their mRNA expression patterns. The population's expression pattern is modeled as the linear combination of mRNA expression from pure samples of cells, allowing reconstruction of the relative proportions of pure cell types in the population. Application of the method, termed expression deconvolution, to yeast grown under varying conditions reveals the population dynamics of the cells during the cell cycle, during the arrest of cells induced by DNA damage and the release of arrest in a cell cycle checkpoint mutant, during sporulation, and following environmental stress. Using expression deconvolution, cell cycle defects are detected and temporally ordered in 146 yeast deletion mutants; six of these defects are independently experimentally validated. Expression deconvolution allows a reinterpretation of the cell cycle dynamics underlying all previous microarray experiments and can be more generally applied to study most forms of cell population dynamics.
Figures
References
-
- Stern, C. D. & Fraser, S. E. (2001) Nat. Cell Biol. 3, E216-E218. - PubMed
-
- Herzenberg, L. A., De Rosa, S. C. & Herzenberg, L. A. (2000) Immunol. Today 21, 383-390. - PubMed
-
- Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K. & Watson, J. D. (1994) Molecular Biology of the Cell (Garland, New York), 3rd Ed.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
