

The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis
- PMID: 12939342
- PMCID: PMC2194184
- DOI: 10.1084/jem.20030116
The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis
Abstract
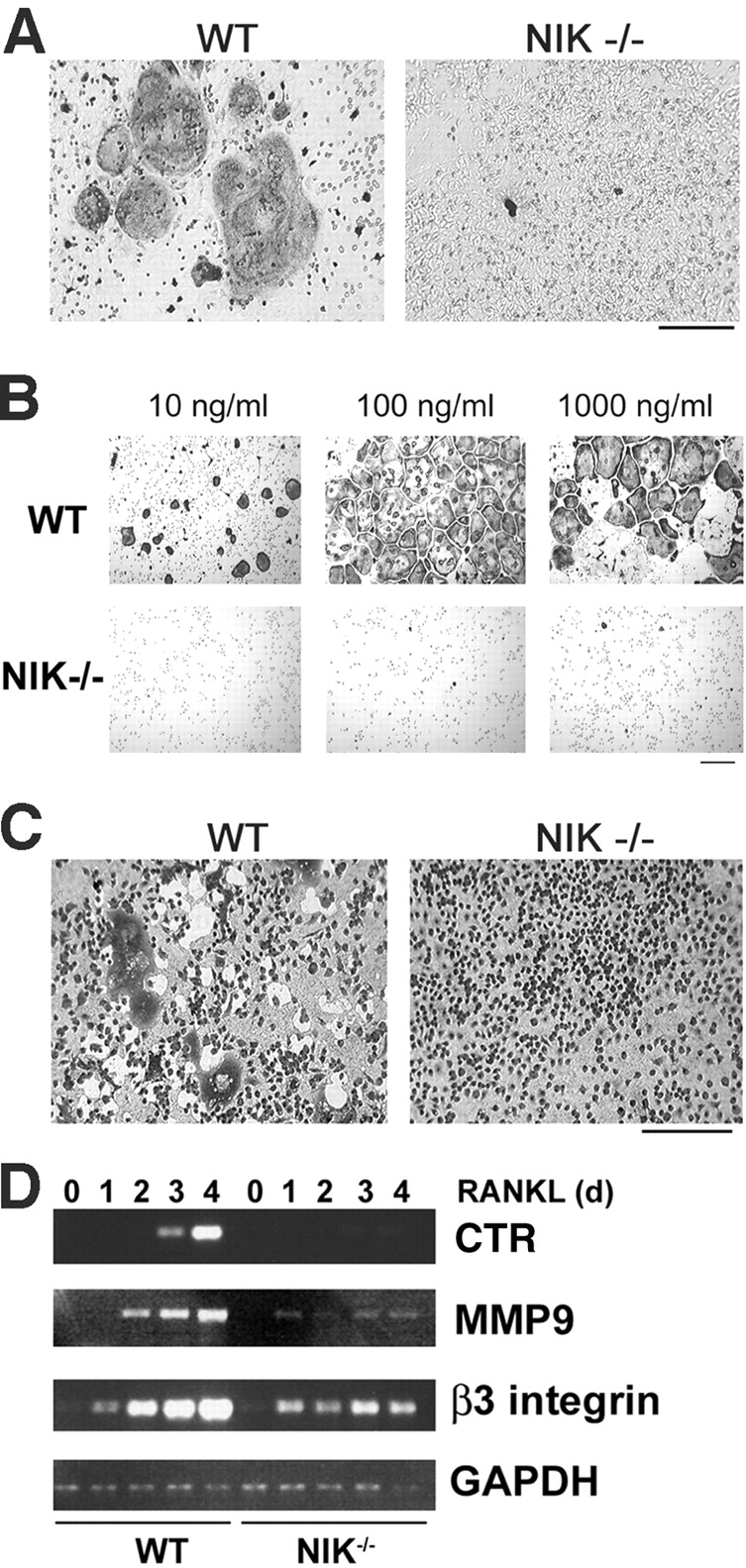
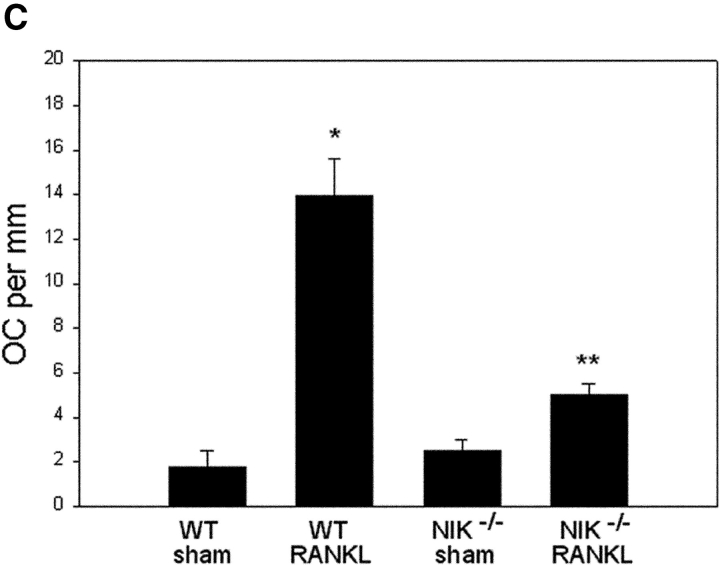
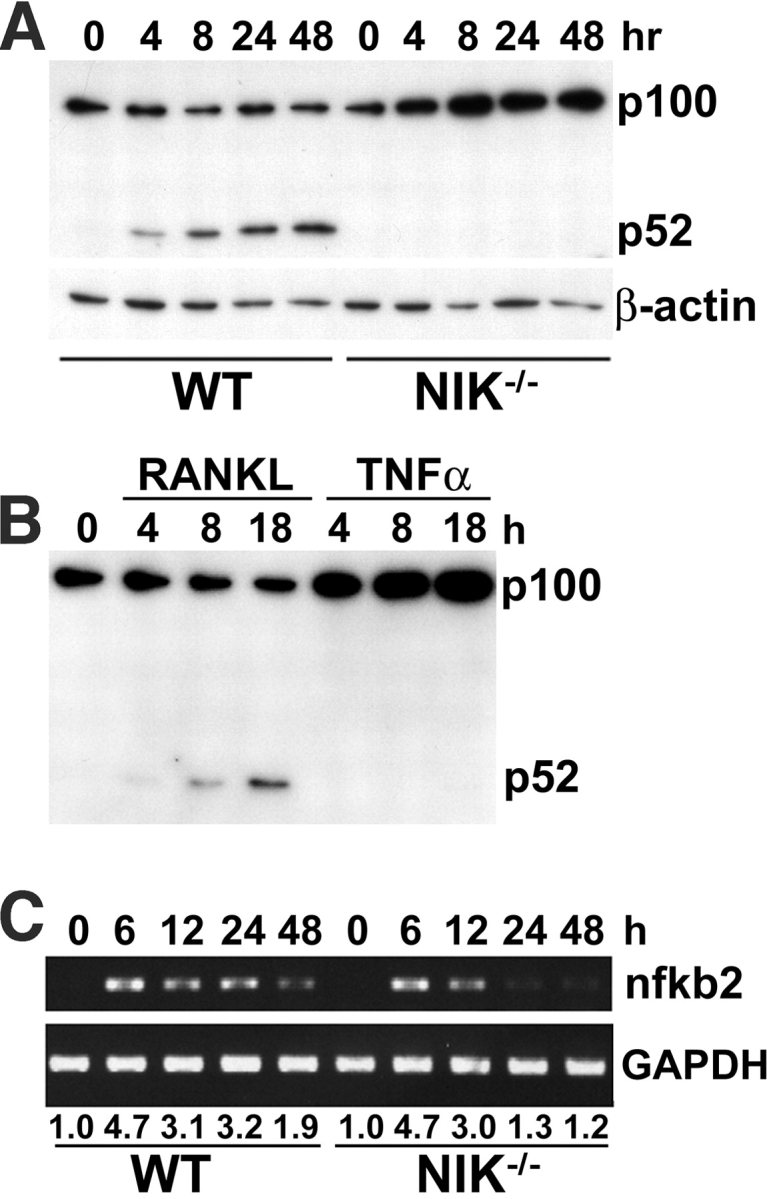
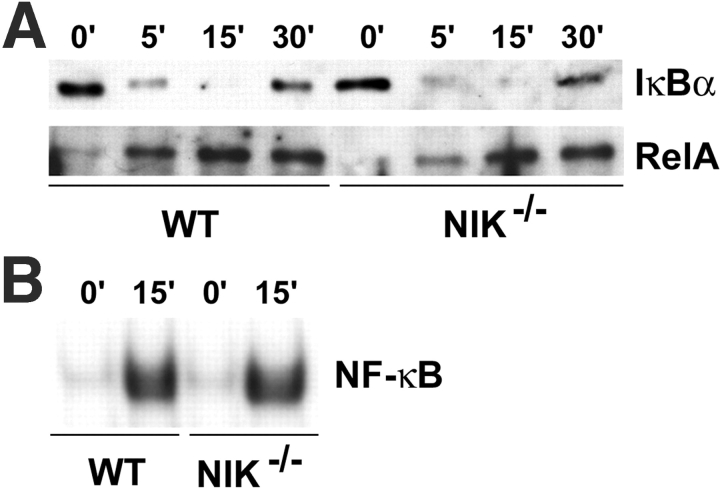
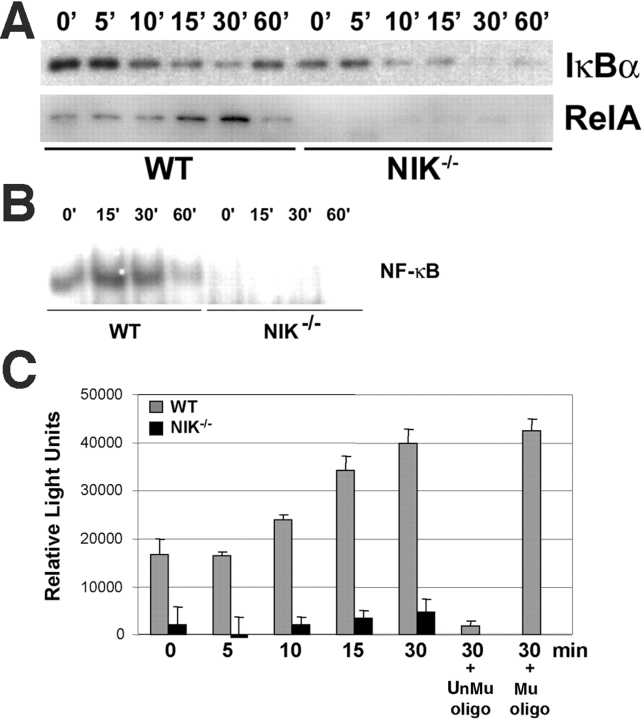
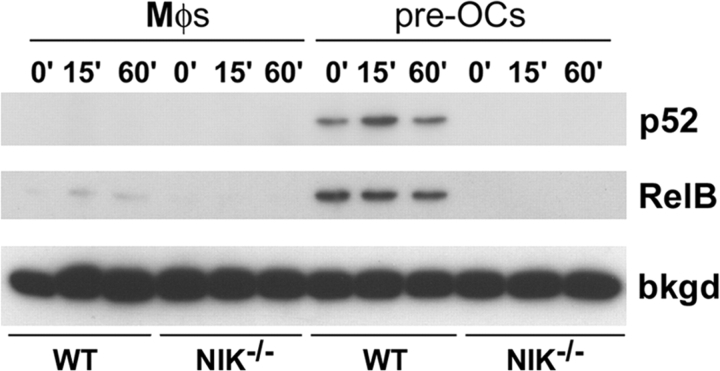
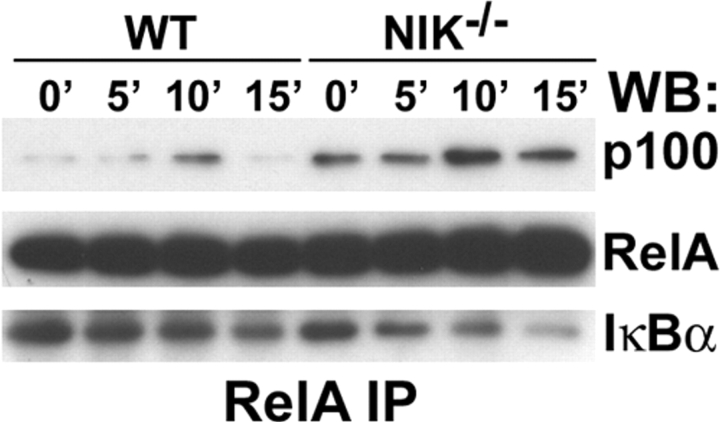
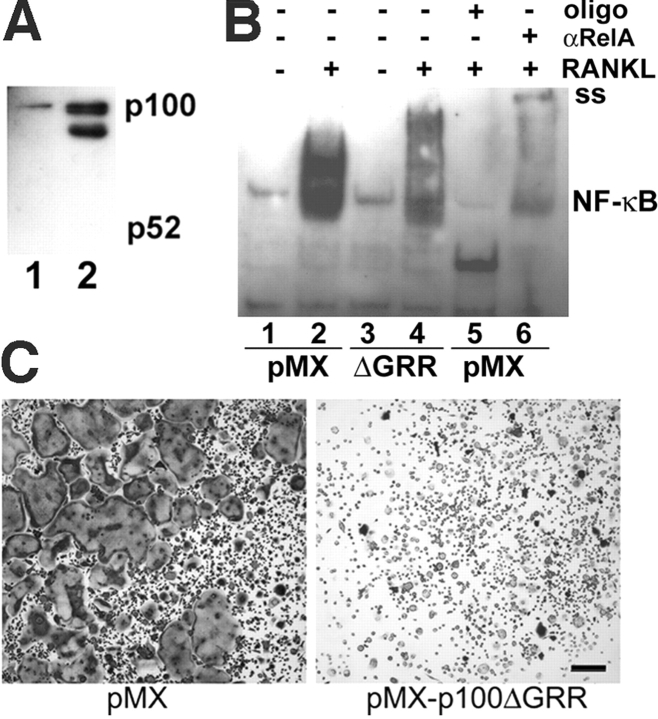
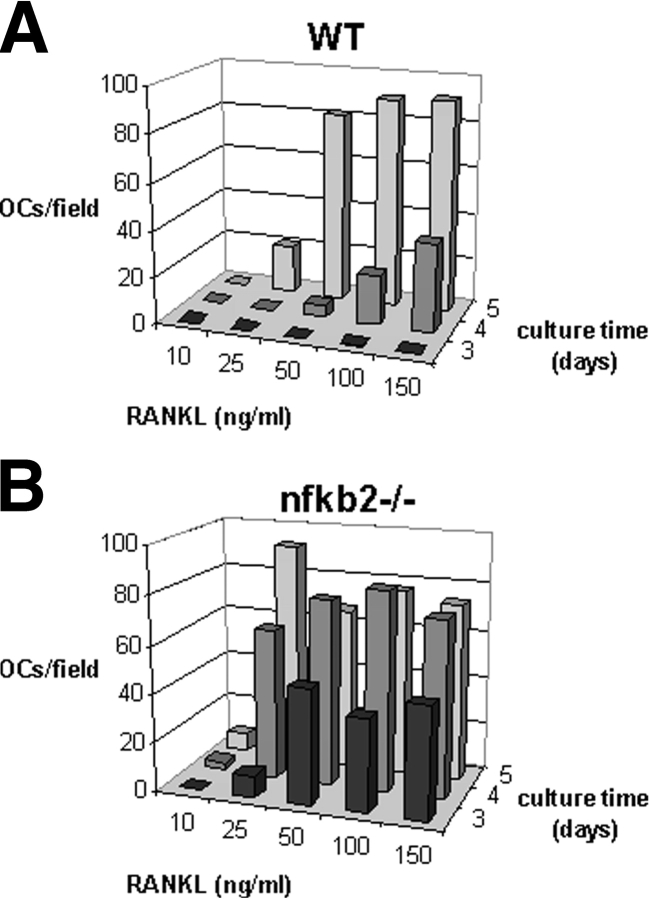
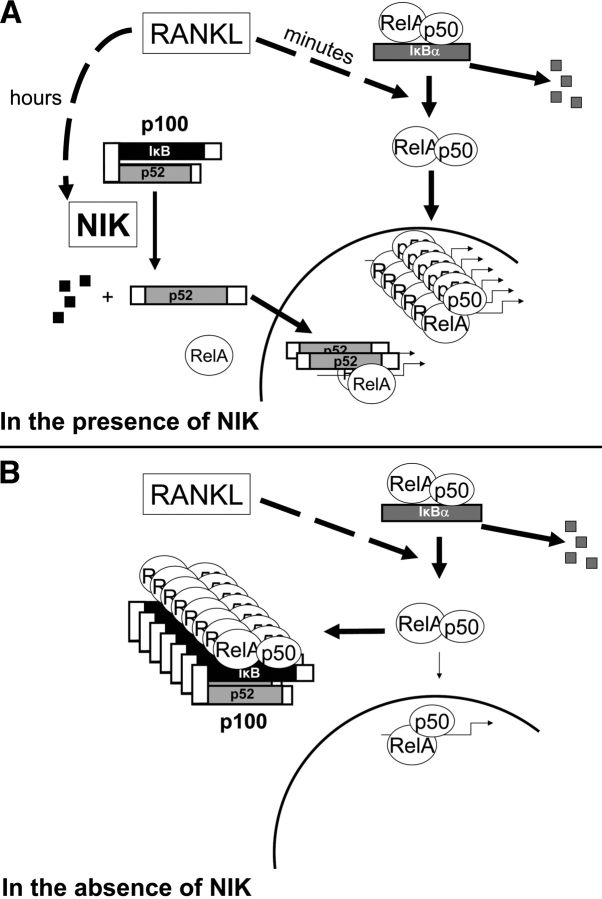
The prototranscription factor p100 represents an intersection of the NF-kappaB and IkappaB families, potentially serving as both the precursor for the active NF-kappaB subunit p52 and as an IkappaB capable of retaining NF-kappaB in the cytoplasm. NF-kappaB-inducing kinase (NIK) controls processing of p100 to generate p52, and thus NIK-deficient mice can be used to examine the biological effects of a failure in such processing. We demonstrate that treatment of wild-type osteoclast precursors with the osteoclastogenic cytokine receptor activator of NF-kappaB ligand (RANKL) increases both expression of p100 and its conversion to p52, resulting in unchanged net levels of p100. In the absence of NIK, p100 expression is increased by RANKL, but its conversion to p52 is blocked, leading to cytosolic accumulation of p100, which, acting as an IkappaB protein, binds NF-kappaB complexes and prevents their nuclear translocation. High levels of unprocessed p100 in osteoclast precursors from NIK-/- mice or a nonprocessable form of the protein in wild-type cells impair RANKL-mediated osteoclastogenesis. Conversely, p100-deficient osteoclast precursors show enhanced sensitivity to RANKL. These data demonstrate a novel, biologically relevant means of regulating NF-kappaB signaling, with upstream control and kinetics distinct from the classical IkappaBalpha pathway.
Figures

References
-
- Teitelbaum, S.L. 2000. Bone resorption by osteoclasts. Science. 289:1504–1508. - PubMed
-
- Ghosh, S., M.J. May, and E.B. Kopp. 1998. NF-κB and REL proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16:225–260. - PubMed
-
- Rothwarf, D.M., and M. Karin. 1999. The NF-κB activation pathway: a paradigm in information transfer from membrane to nucleus. Sci. STKE. 1999:RE1. - PubMed
-
- Palombella, V.J., O.J. Rando, A.L. Goldberg, and T. Maniatis. 1994. The ubiquitin-proteasome pathway is required for processing of the NF-κB1 precursor protein and the activation of NF-κB. Cell. 78:773–785. - PubMed
-
- Heusch, M., L. Lin, R. Geleziunas, and W.C. Greene. 1999. The generation of nfkb2 p52: mechanism and efficiency. Oncogene. 16:6201–6208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AR 48853/AR/NIAMS NIH HHS/United States
- R01 AR 32788/AR/NIAMS NIH HHS/United States
- 5T32 DK 07120/DK/NIDDK NIH HHS/United States
- R01 AR046523/AR/NIAMS NIH HHS/United States
- K08 AR047846/AR/NIAMS NIH HHS/United States
- R01 AR 48812/AR/NIAMS NIH HHS/United States
- R01 CA 43509/CA/NCI NIH HHS/United States
- R01 AR046852/AR/NIAMS NIH HHS/United States
- R01 AR 46852/AR/NIAMS NIH HHS/United States
- R01 AR032788/AR/NIAMS NIH HHS/United States
- R01 AR048812/AR/NIAMS NIH HHS/United States
- R01 AR048853/AR/NIAMS NIH HHS/United States
- T32 DK007120/DK/NIDDK NIH HHS/United States
- P30 AR 48335/AR/NIAMS NIH HHS/United States
- K08 AR 47846/AR/NIAMS NIH HHS/United States
- R01 AR 46523/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
