

Similarity of force-induced unfolding of apomyoglobin to its chemical-induced unfolding: an atomistic molecular dynamics simulation approach
- PMID: 12944267
- PMCID: PMC1303326
- DOI: 10.1016/S0006-3495(03)74582-2
Similarity of force-induced unfolding of apomyoglobin to its chemical-induced unfolding: an atomistic molecular dynamics simulation approach
Abstract
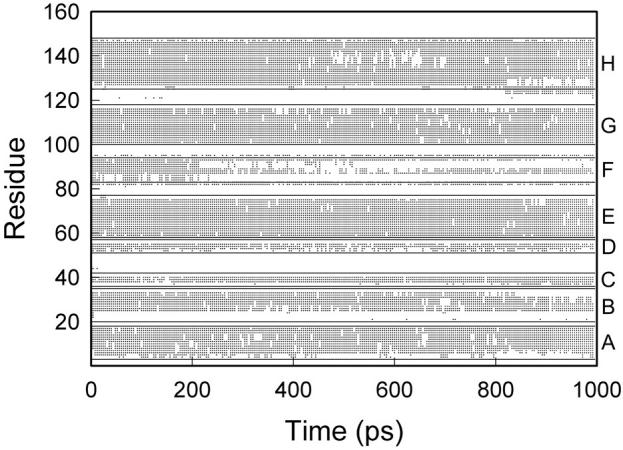
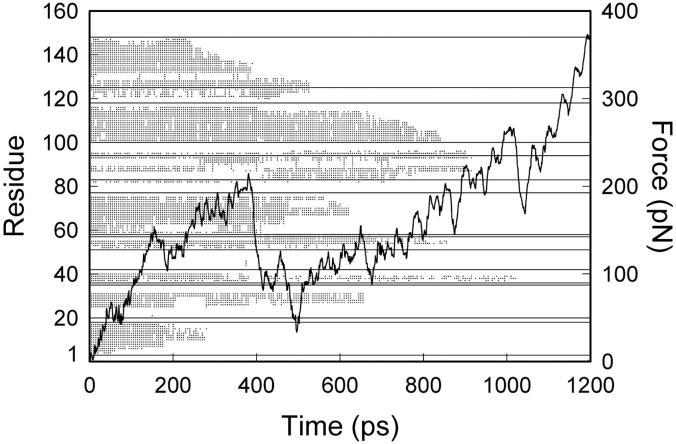
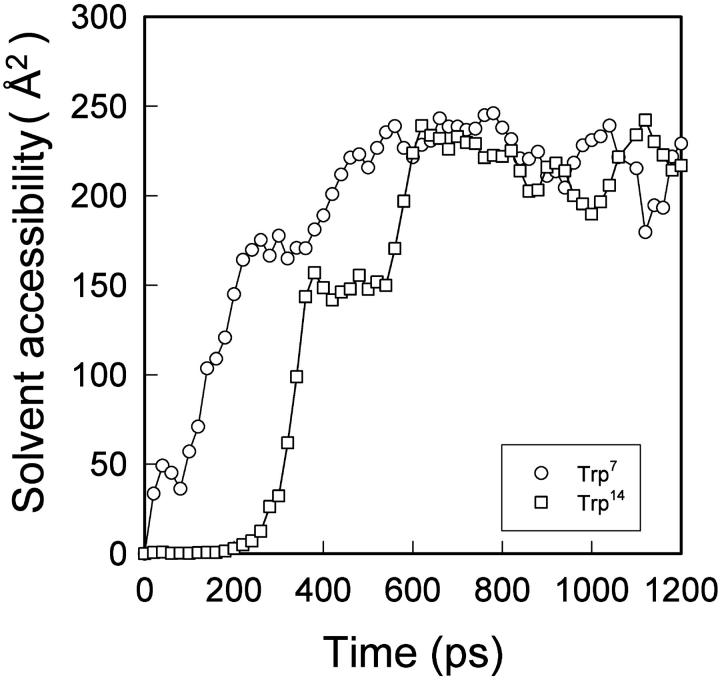
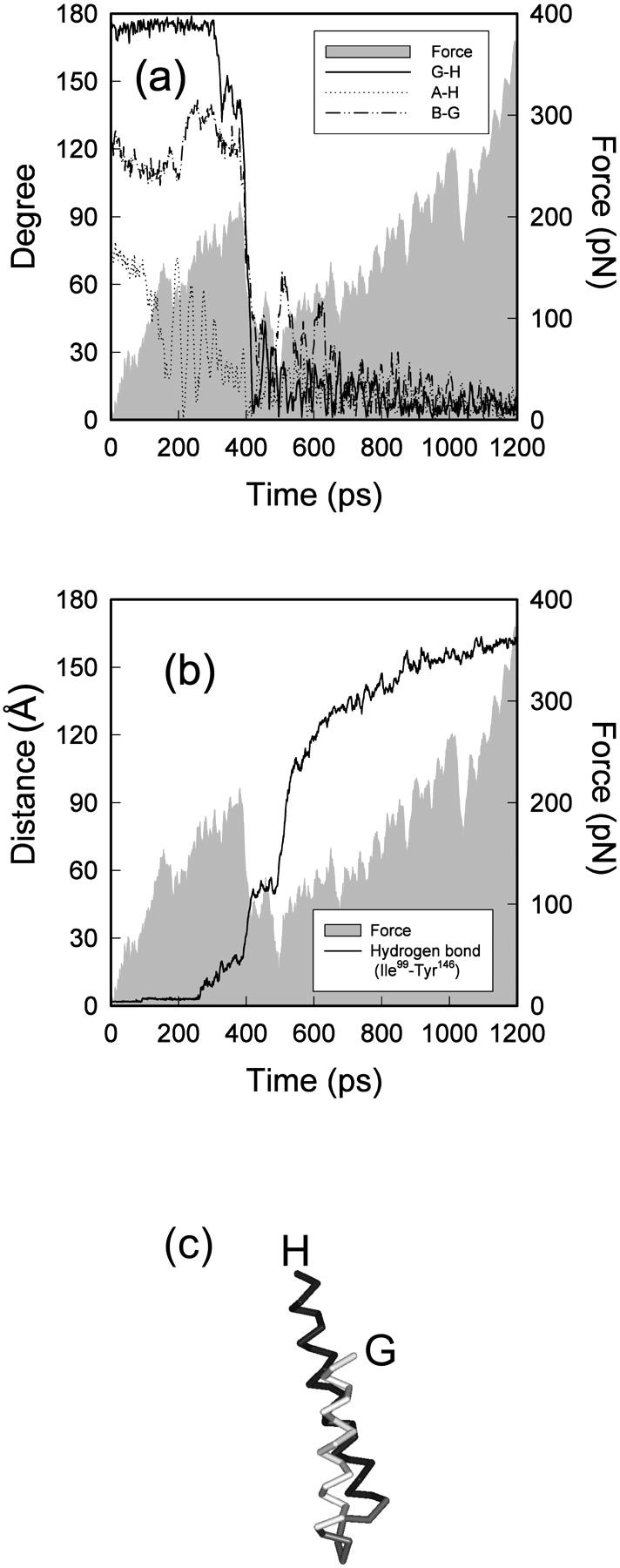
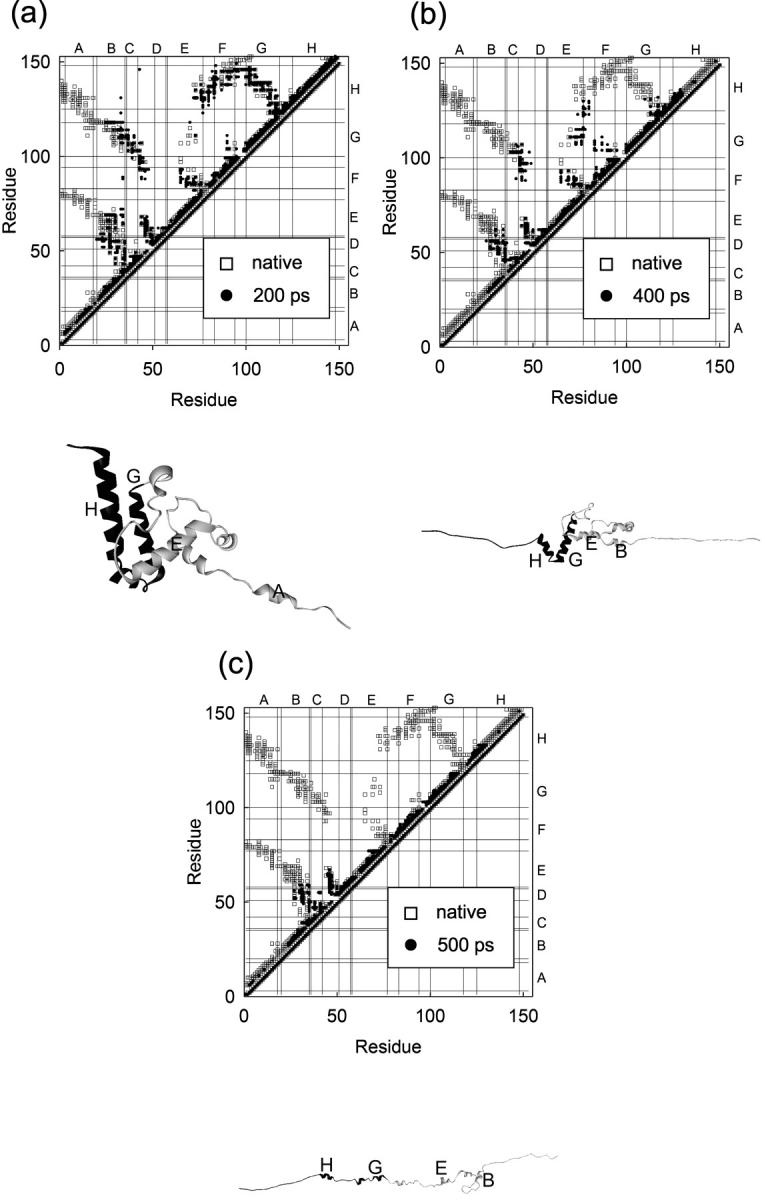
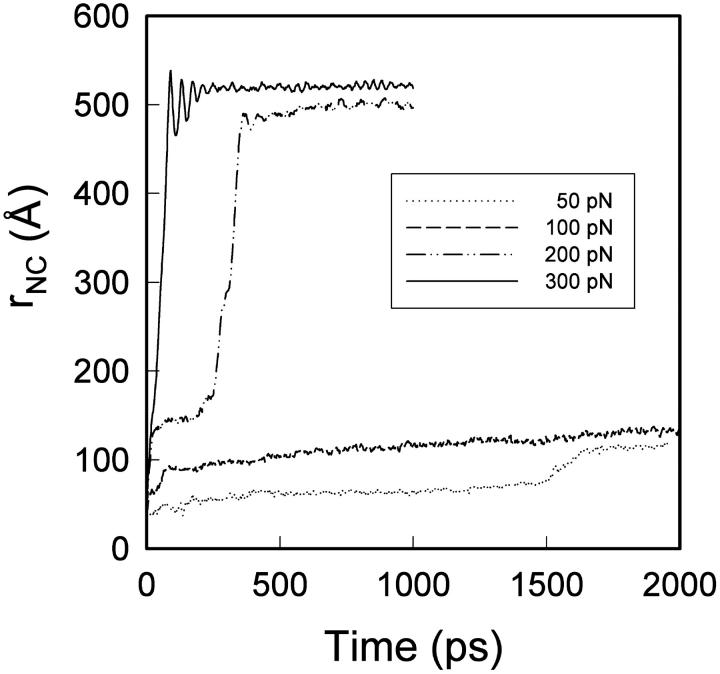
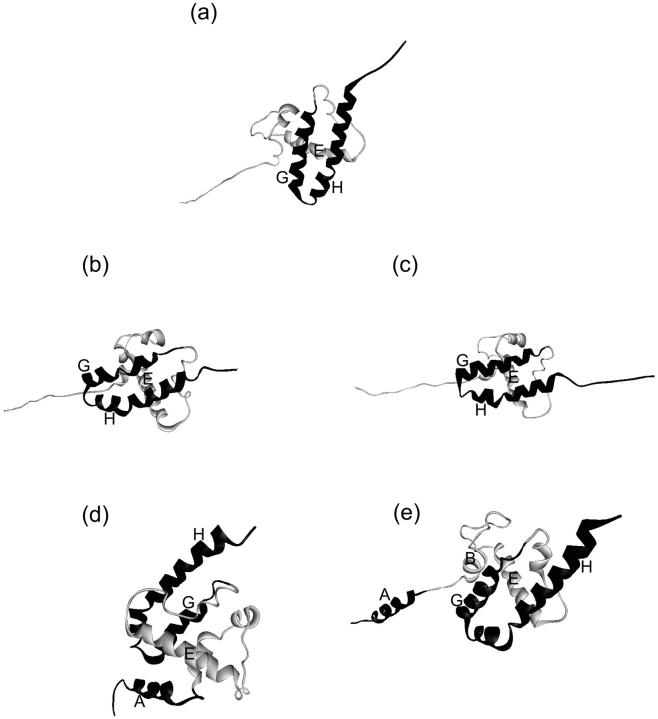
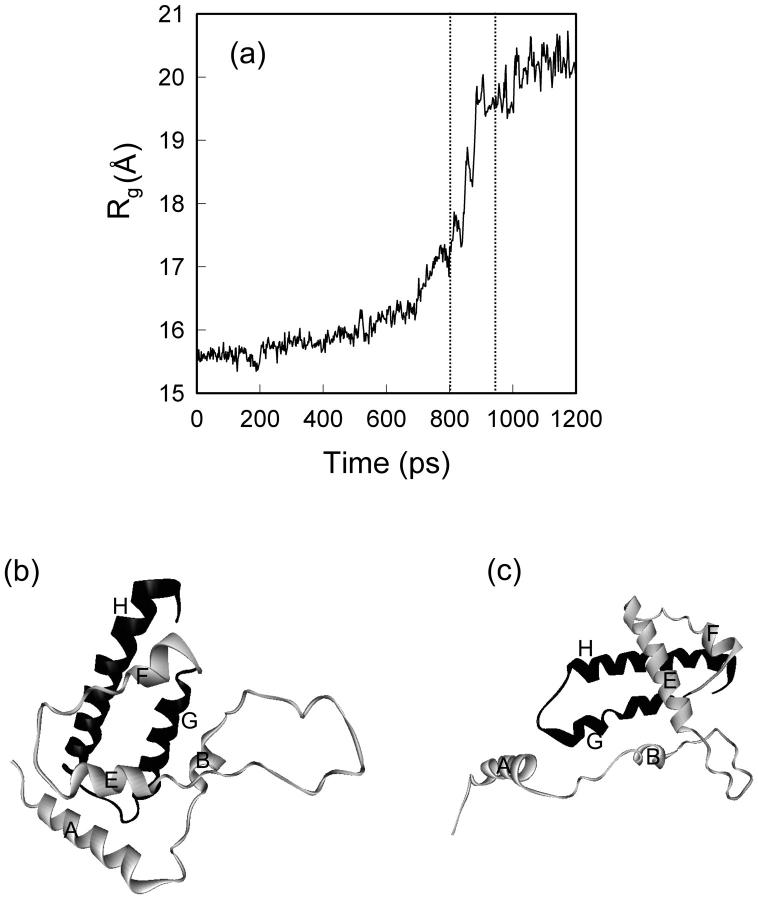
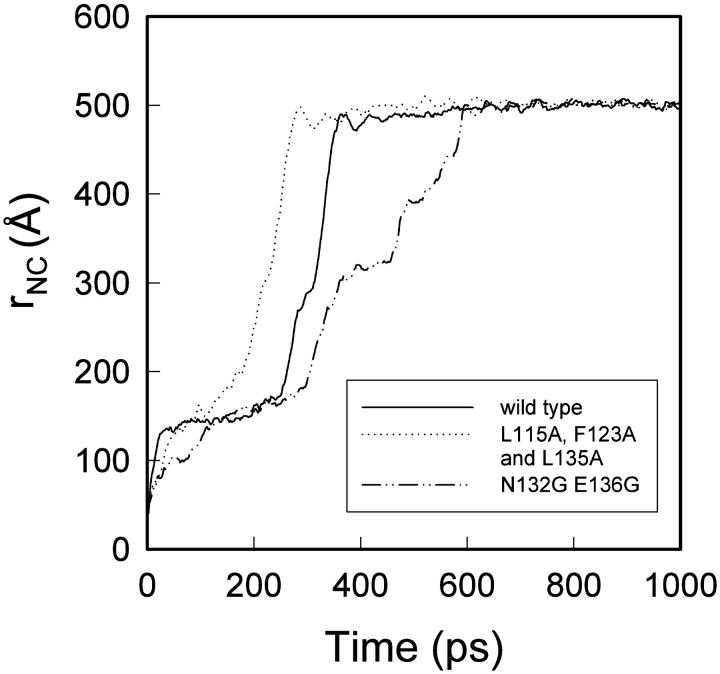
We have compared force-induced unfolding with traditional unfolding methods using apomyoglobin as a model protein. Using molecular dynamics simulation, we have investigated the structural stability as a function of the degree of mechanical perturbation. Both anisotropic perturbation by stretching two terminal atoms and isotropic perturbation by increasing the radius of gyration of the protein show the same key event of force-induced unfolding. Our primary results show that the native structure of apomyoglobin becomes destabilized against the mechanical perturbation as soon as the interhelical packing between the G and H helices is broken, suggesting that our simulation results share a common feature with the experimental observation that the interhelical contact is more important for the folding of apomyoglobin than the stability of individual helices. This finding is further confirmed by simulating both helix destabilizing and interhelical packing destabilizing mutants.
Figures

References
-
- Barrick, D., and R. L. Baldwin. 1993. Three-state analysis of sperm whale apomyoglobin folding. Biochemistry. 32:3790–3796. - PubMed
-
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization and dynamics calculations. J. Comput. Chem. 4:187–217.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
