

A tree-based algorithm for determining the effects of solvation on the structure of salivary gland tripeptide NH3+-D-PHE-D-GLU-GLY-COO-
- PMID: 12944268
- PMCID: PMC1303327
- DOI: 10.1016/S0006-3495(03)74583-4
A tree-based algorithm for determining the effects of solvation on the structure of salivary gland tripeptide NH3+-D-PHE-D-GLU-GLY-COO-
Abstract
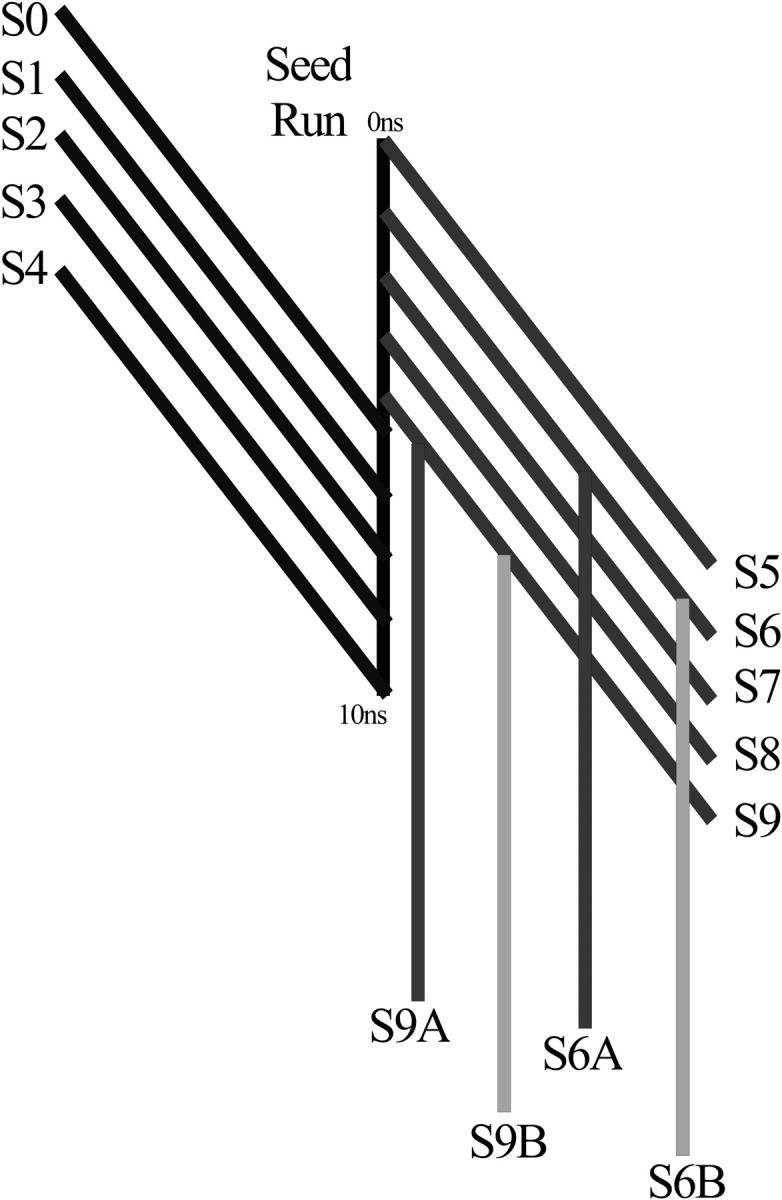
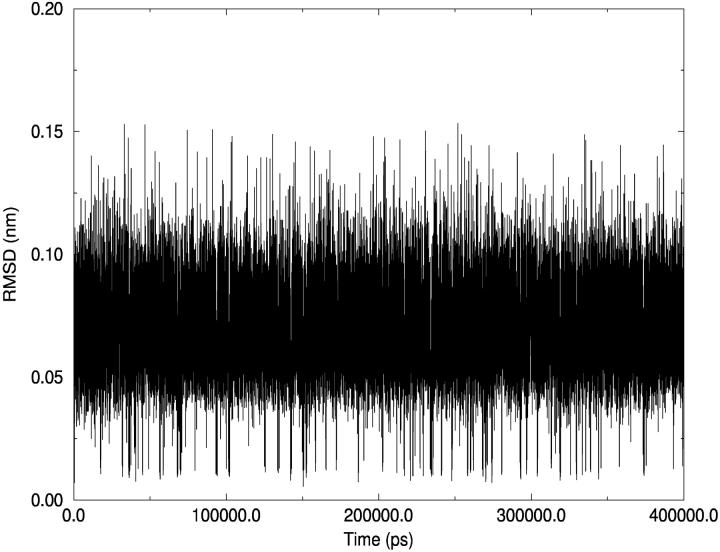
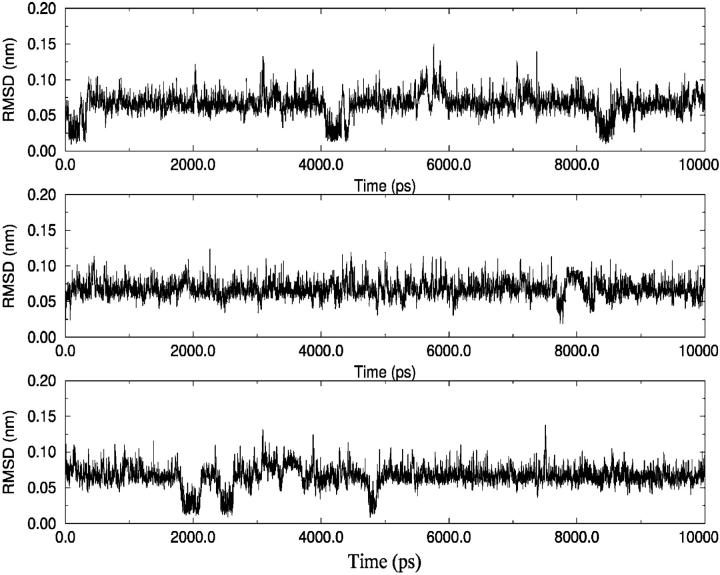
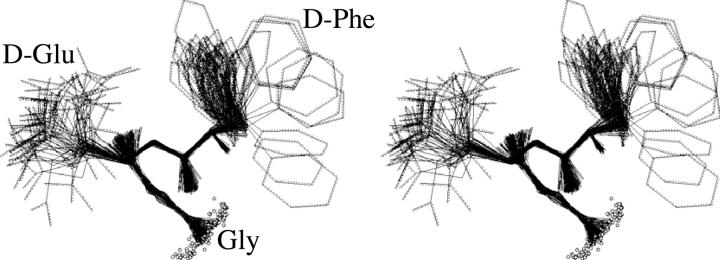
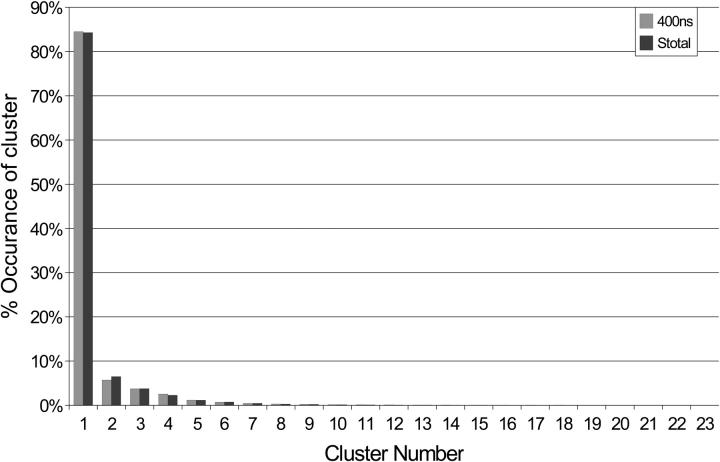
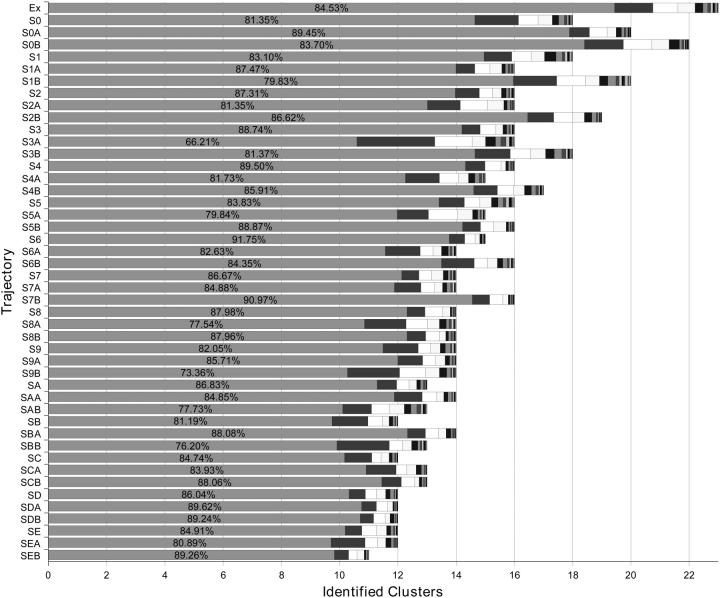
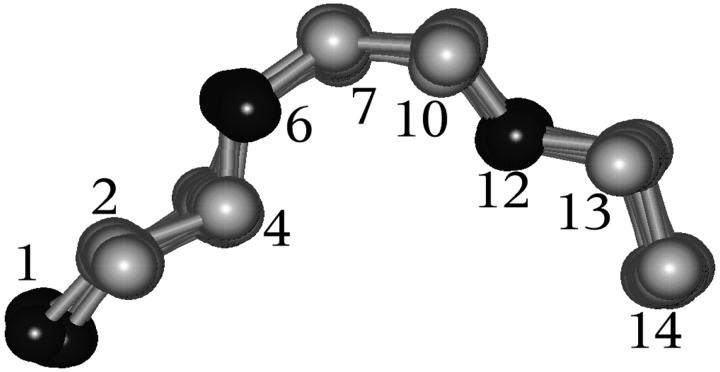
A D-enantiomeric analog of the submandibular gland rat-1 tripeptide FEG (Seq: NH(3)(+)-Phe-Glu-Gly-COO(-)) called feG (Seq: NH(3)(+)-D-Phe-D-Glu-Gly-COO(-)) was examined by molecular dynamics simulations in water. Previous in vacuo simulations suggested a conformation consisting predominantly of interactions between the Phe side chain and glutamyl-carboxyl group and a carboxyl/amino termini interaction. The solvated peptide was simulated using two approaches which were compared-a single 400-ns simulation and a "simulation tree." The "tree" approach utilized 45 10-ns simulations with different conformations used as initial structures for given trajectories. We demonstrate that multiple short duration simulations are able to describe the same conformational space as that described by longer simulations. Furthermore, previously described in vacuo interactions were confirmed with amendments: the previously described head-to-tail arrangement of the amino and carboxyl termini, was not observed; the interaction between the glutamyl carboxyl and Phe side chain describes only one of a continuum of conformations present wherein the aromatic residue remains in close proximity to the glutamyl carbonyl group, and also interacts with either of the two available carboxyl groups. Finally, utilizing only two separate 10-ns trajectories, we were able to better describe the conformational space than a single 60-ns trajectory, realizing a threefold decrease in the computational complexity of the problem.
Figures
References
-
- Angeletti, L. R., U. Agrimi, C. Curia, D. French, and R. Mariani-Constantini. 1992. Healing rituals and sacred serpents. Lancet. 340:223–225. - PubMed
-
- Balsera, M. A., W. Wriggers, Y. Oono, and K. Schulten. 1996. Principal component analysis and long time protein dynamics. J. Phys. Chem. 100:2567–2572.
-
- Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 91:43–56.
-
- Burgi, R., X. Daura, A. Mark, M. Bellanda, S. Mammi, E. Peggion, and W. van Gunsteren. 2001. Folding study of an Aib-rich peptide in DMSO by molecular dynamics simulations. J. Pept. Res. 57:107–118. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
