

Position specific variation in the rate of evolution in transcription factor binding sites
- PMID: 12946282
- PMCID: PMC212491
- DOI: 10.1186/1471-2148-3-19
Position specific variation in the rate of evolution in transcription factor binding sites
Abstract
Background: The binding sites of sequence specific transcription factors are an important and relatively well-understood class of functional non-coding DNAs. Although a wide variety of experimental and computational methods have been developed to characterize transcription factor binding sites, they remain difficult to identify. Comparison of non-coding DNA from related species has shown considerable promise in identifying these functional non-coding sequences, even though relatively little is known about their evolution.
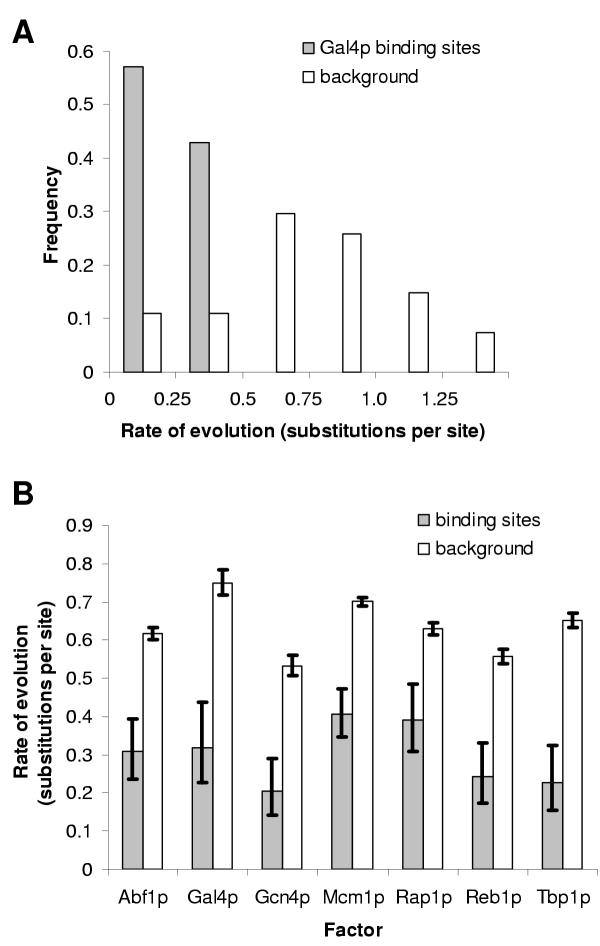
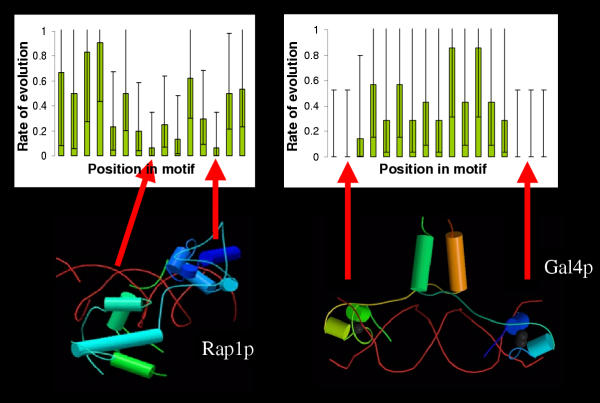
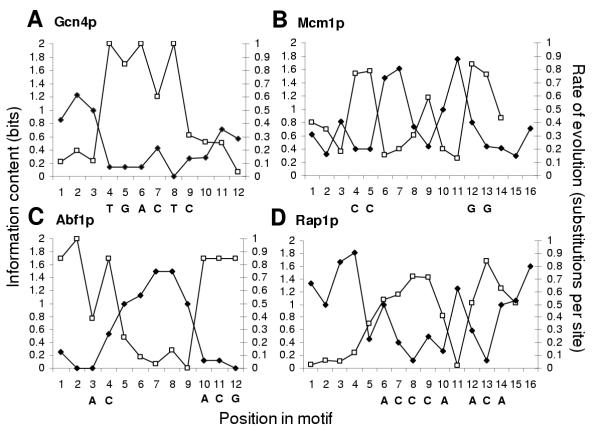
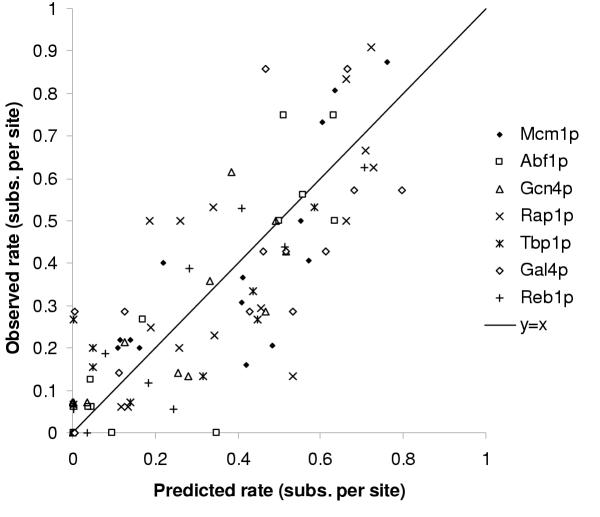
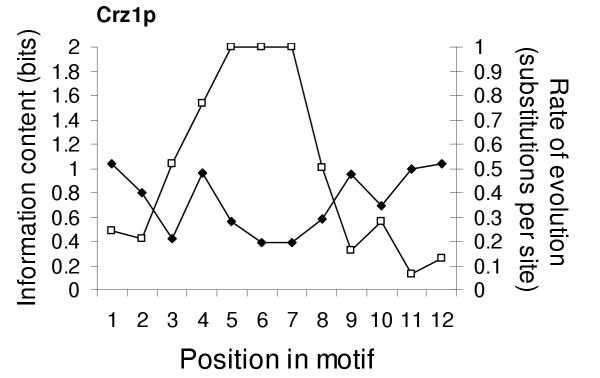
Results: Here we analyse the genome sequences of the budding yeasts Saccharomyces cerevisiae, S. bayanus, S. paradoxus and S. mikatae to study the evolution of transcription factor binding sites. As expected, we find that both experimentally characterized and computationally predicted binding sites evolve slower than surrounding sequence, consistent with the hypothesis that they are under purifying selection. We also observe position-specific variation in the rate of evolution within binding sites. We find that the position-specific rate of evolution is positively correlated with degeneracy among binding sites within S. cerevisiae. We test theoretical predictions for the rate of evolution at positions where the base frequencies deviate from background due to purifying selection and find reasonable agreement with the observed rates of evolution. Finally, we show how the evolutionary characteristics of real binding motifs can be used to distinguish them from artefacts of computational motif finding algorithms.
Conclusion: As has been observed for protein sequences, the rate of evolution in transcription factor binding sites varies with position, suggesting that some regions are under stronger functional constraint than others. This variation likely reflects the varying importance of different positions in the formation of the protein-DNA complex. The characterization of the pattern of evolution in known binding sites will likely contribute to the effective use of comparative sequence data in the identification of transcription factor binding sites and is an important step toward understanding the evolution of functional non-coding DNA.
Figures
References
-
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. - PubMed
-
- Bailey TL, Elkan C. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. AAAI Press, Menlo Park, California; 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers; pp. 28–36. - PubMed
-
- Eskin E, Pevzner PA. Finding composite regulatory patterns in DNA sequences. Bioinformatics. 2002;18:S354–363. - PubMed
-
- Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol. 2002;20:835–839. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
