

Review
doi: 10.1038/nrd1176.
Recent advances in the discovery and delivery of vaccine adjuvants
Affiliations
- PMID: 12951579
- PMCID: PMC7097252
- DOI: 10.1038/nrd1176
Item in Clipboard
Review
Recent advances in the discovery and delivery of vaccine adjuvants
Nat Rev Drug Discov.
2003 Sep.
Abstract
Adjuvant design has historically had a touch of alchemy at its heart due to its reliance on the complex biology of innate immune activation. However, a new mechanistic understanding of innate immunity, combined with new adjuvant and delivery platforms for exploiting this knowledge, has led to significant advances recently. Although many challenges remain, the field is moving rapidly and the proper tools and methodologies are in place for the use of traditional drug discovery engines in guiding the development of vaccine adjuvants. In this review, we outline the current trends in immune potentiator, delivery system and adjuvant design that will shape the vaccines of the future.
Figures

It is well known that subunit vaccines elicit more potent and durable antigen-specific immunity if combined with an adjuvant. The in vivo adjuvant effect can be divided into two principal components: delivery and immune potentiation. Delivery systems localize antigens and target them to the appropriate cell types of the innate immune system. Delivery can also be optimized for immune potentiator targeting. Immune potentiators directly activate innate immune cells providing the pro-inflammatory context for antigen recognition. Antigens provide the specific pathogen epitopes necessary to generate long-lived immunological memory. These three components are intrinsic to naturally occurring infections and whole-cell vaccines, whereas they must be combined in subunit vaccine formulations.

a | The division of labour between the innate and adaptive (antigen-specific) immune responses on first encounter with a vaccine or infectious agent maximizes the two mutually exclusive priorities of host defence (speed and specificity). The equal importance of these competing goals can be seen when they are merged in the remarkably efficient memory response. Generating rapid, specific and durable memory responses is the goal of vaccination. In general, immune potentiators and delivery systems target the innate arm, whereas vaccine antigens drive pathogen-specific responses by B and T cells. b | The arrows indicate cytokine networks and cross-talk between regulatory cells of both innate and antigen-specific immune systems, which are central to the initiation and amplification of specific T- and B-cell responses. Key cytokines that both amplify and steer effector cell generation include interleukin-2 (IL-2), IL-4, IL-12, type I and II interferons (IFN) and other pro-inflammatory mediators (for example, tumour-necrosis factor (TNF)). c | In the effector and memory stages, the elicited cell-mediated and humoral mechanisms potently and specifically target the pathogen and/or infected cells for destruction. Abs, antibodies; ADCC, antibody-dependent cell-mediated cytotoxicity; CTL, cytotoxic T lymphocyte; Imm-Pot, immune potentiator; mDC/pDC, myeloid/plasmacytoid dendritic cell; mono, monocyte; MΦ, macrophage; NC, natural cytotoxicity; NK, natural killer cell; NKT, natural killer T cell; Phag, phagocytosis; PMN, polymorphonuclear leukocyte (neutrophil); TH1, helper T cell 1 (cell-mediated immunity); TH2, helper T cell 2 (humoral immunity).
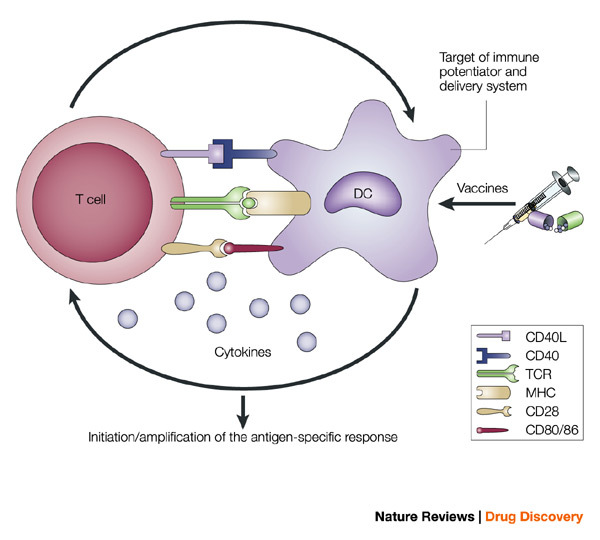
The intimate interactions (cognate recognition) between antigen-presenting cells (APCs) and antigen-specific T cells initiate and amplify pathogen-specific responses. A dendritic cell (DC) is depicted here. The key interaction is driven by the recognition of antigenic peptide–major histocompatibility complex (MHC) dimers by T cells bearing T-cell receptors (TCRs) with high affinity for the complex. However, this signal alone is not sufficient for initiation and amplification of specific T-cell responses. Co-stimulatory signals (that is, CD28 recognition of CD80/CD86) and the production of pro-inflammatory cytokines, provide the 'infectious context' by which the full activation of antigen-specific T cells is achieved. The expression of co-stimulatory molecules and cytokines by APCs is tightly regulated and induced only when the APC encounters antigens associated with pathogen-associated molecular patterns. From the perspective of adjuvant design, the APC is a high-priority target. Delivery systems increase antigen uptake and presentation, and can also target immune potentiators more efficiently to APCs. Immune potentiators induce co-stimulatory signals and cytokine production, and the antigens select the highly specific T cells leading to the initiation and amplification of antigen-specific immunity. CD40L, CD40 ligand.
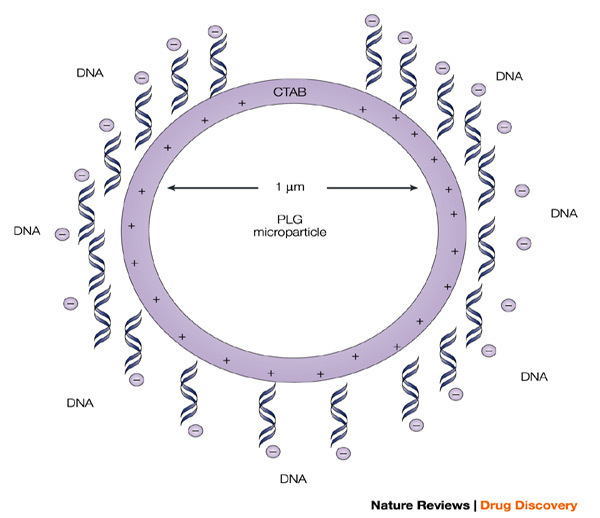
The microparticle is cationic because of the presence of the cationic detergent hexadecyltrimethyl ammonium bromide (CTAB), which binds to the surface during microparticle preparation, and allows the efficient adsorption of poly-anionic plasmids. This approach serves to significantly enhance the potency of the antigens encoded by the adsorbed DNA vaccines.
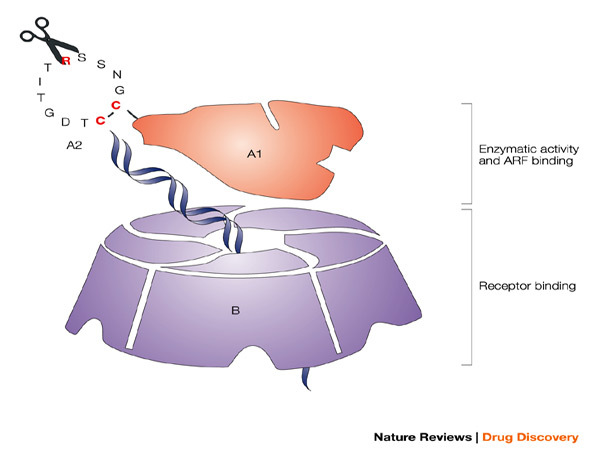
The potent mucosal immunogen and adjuvant heat-labile enterotoxin from Escherichia coli (LT), with the A and B subunits and their structural and functional activities, is shown. The B oligomer is a pentameric molecule of 55 kDa, which contains five identical polypeptide monomers and is responsible for binding to mucosal epithelial cells. The A1 subunit comprises a globular structure, which is enzymatically active and mediates ADP-ribosylation, resulting in permanent activation of adenylate cyclase, abnormal intracellular accumulation of cAMP and massive fluid loss, resulting in diarrhoea. The A1 subunit is linked to the B oligomer with the long helical A2 subunit, which must be proteolytically cleaved in addition to a disulphide being reduced to release and activate the A1 subunit. The ADP-ribosyltransferase activity of the A1 subunit is enhanced by interaction with intracellular ADP-ribosylation factors (ARF). Various groups have attempted to generate potent but safe mucosal adjuvants from LT, through a variety of genetic manipulations. Chiron scientists have focused on modifying or eliminating the enzymatic activity of LT (ADP-ribosylation) through single amino-acid substitutions in the active site of A1 (LTK63 and LTR72), while an alternative approach pursued by others was to modify the ability of the A1 subunit to be proteolytically cleaved from A2 by trypsin (LTR192G).

In vitro toxicity on Y1 adrenal cells is shown for heat-labile enterotoxin (LT) wild type (LTwt), for the mutants generated by Chiron, which have modifications in the enzymatic active site of the A1 subunit (LTK63 and LTR72), and for an alternative mutant, LTR192G, which is modified in its ability to be enzymatically cleaved and activated by trypsin. LTK63 has no enzyme activity in the A1 subunit and shows no toxicity on Y1 cells, whereas LTR72 has residual enzyme activity (about 0.6% of LTwt) and shows residual toxicity, but is 100,000-fold less toxic than LTwt. By contrast, LTR192G shows only a marginal reduction in toxicity. Similar results were obtained for in vivo toxicity in the standard rabbit ileal loop model, which shows significant fluid accumulation with LTwt. In contrast, LTK63 shows no fluid accumulation, even at the highest dose tested (1 mg), whereas LTR72 shows reduced fluid accumulation and the requirement for a higher dose to trigger this effect. LTR192G shows high levels of fluid accumulation, even at low doses, similar to LTwt, probably because there are alternative enzymes present that can cleave the molecule, in addition to trypsin. For further details on experimental methodology for these studies see Refs ,.
References
-
- WHO World Health Report (2002).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
