

A transcriptional regulatory cascade that controls left/right asymmetry in chemosensory neurons of C. elegans
- PMID: 12952888
- PMCID: PMC196454
- DOI: 10.1101/gad.1117903
A transcriptional regulatory cascade that controls left/right asymmetry in chemosensory neurons of C. elegans
Abstract
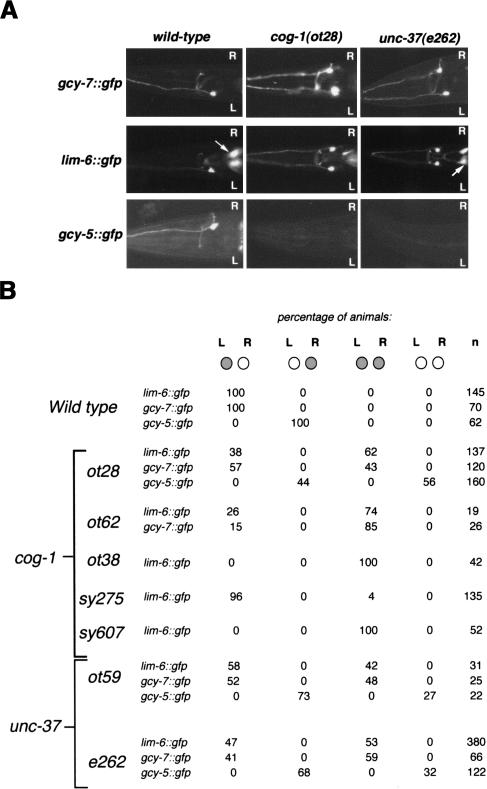
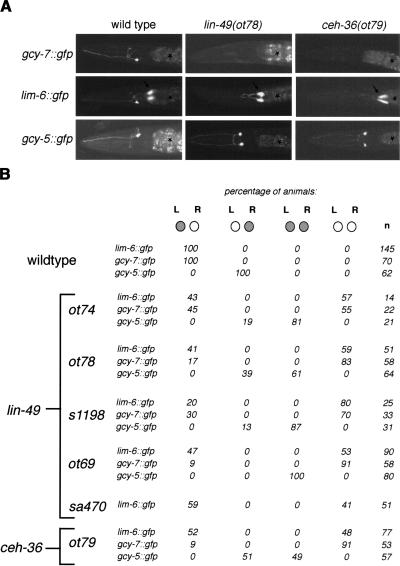
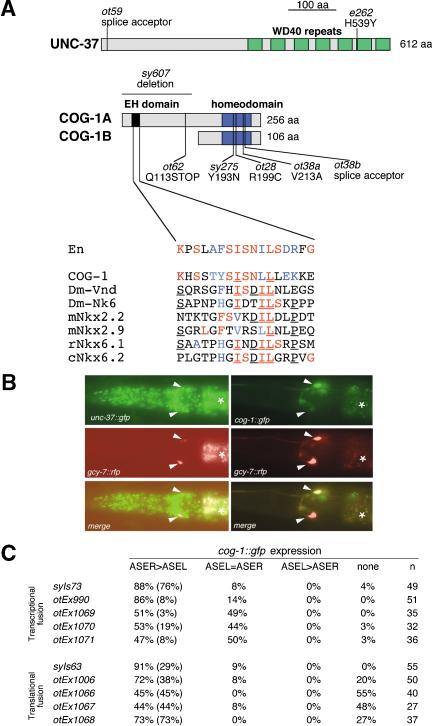
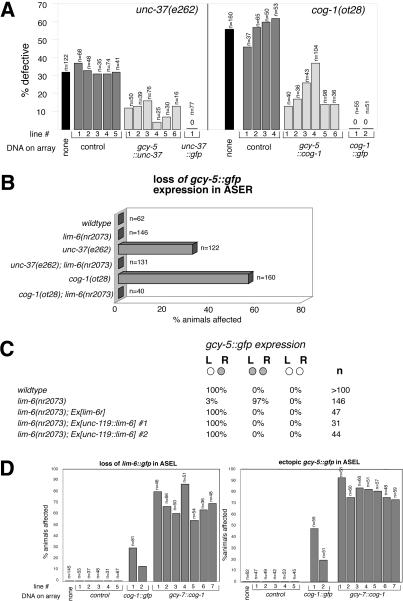
The molecular mechanisms of differential pattern formation along the left/right (L/R) axis in the nervous system are poorly understood. The nervous system of the nematode Caenorhabditis elegans displays several examples of L/R asymmetry, including the directional asymmetry displayed by the two ASE taste receptor neurons, ASE left (ASEL) and ASE right (ASER). Although bilaterally symmetric in regard to all known morphological criteria, these two neurons display distinct chemosensory capacities that correlate with the L/R asymmetric expression of three putative sensory receptor genes, gcy-5, expressed only in ASER, and gcy-6 and gcy-7, expressed only in ASEL. In order to understand the genetic basis of L/R asymmetry establishment, we screened for mutants in which patterns of asymmetric gcy gene expression are disrupted, and we identified a cascade of several symmetrically and asymmetrically expressed transcription factors that are sequentially required to restrict gcy gene expression to either the left or right ASE cell. These factors include the zinc finger transcription factor che-1; the homeobox genes cog-1, ceh-36, and lim-6; and the transcriptional cofactors unc-37/Groucho and lin-49. Specific features of this regulatory hierarchy are sequentially acting repressive interactions and the finely balanced activity of antagonizing positive and negative regulatory factors. A key trigger for asymmetry is the L/R differential expression of the Nkx6-type COG-1 homeodomain protein. Our studies have thus identified transcriptional mediators of a putative L/R-asymmetric signaling event and suggest that vertebrate homologs of these proteins may have similar functions in regulating vertebrate brain asymmetries.
Figures





References
-
- Aasland R., Gibson, T.J., and Stewart, A.F. 1995. The PHD finger: Implications for chromatin-mediated transcriptional regulation. Trends Biochem. Sci. 20: 56–59. - PubMed
-
- Bargmann C.I., Hartwieg, E., and Horvitz, H.R. 1993. Odorant-selective genes and neurons mediate olfaction in C. elegans. Cell 74: 515–527. - PubMed
-
- Chamberlin H.M. and Thomas, J.H. 2000. The bromodomain protein LIN-49 and trithorax-related protein LIN-59 affect development and gene expression in Caenorhabditis elegans. Development 127: 713–723. - PubMed
-
- Concha M.L., Burdine, R.D., Russell, C., Schier, A.F., and Wilson, S.W. 2000. A nodal signaling pathway regulates the laterality of neuroanatomical asymmetries in the zebrafish forebrain. Neuron 28: 399–409. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous