

Integration of cell attachment, cytoskeletal localization, and signaling by integrin-linked kinase (ILK), CH-ILKBP, and the tumor suppressor PTEN
- PMID: 12960424
- PMCID: PMC284786
- DOI: 10.1091/mbc.e03-05-0308
Integration of cell attachment, cytoskeletal localization, and signaling by integrin-linked kinase (ILK), CH-ILKBP, and the tumor suppressor PTEN
Abstract
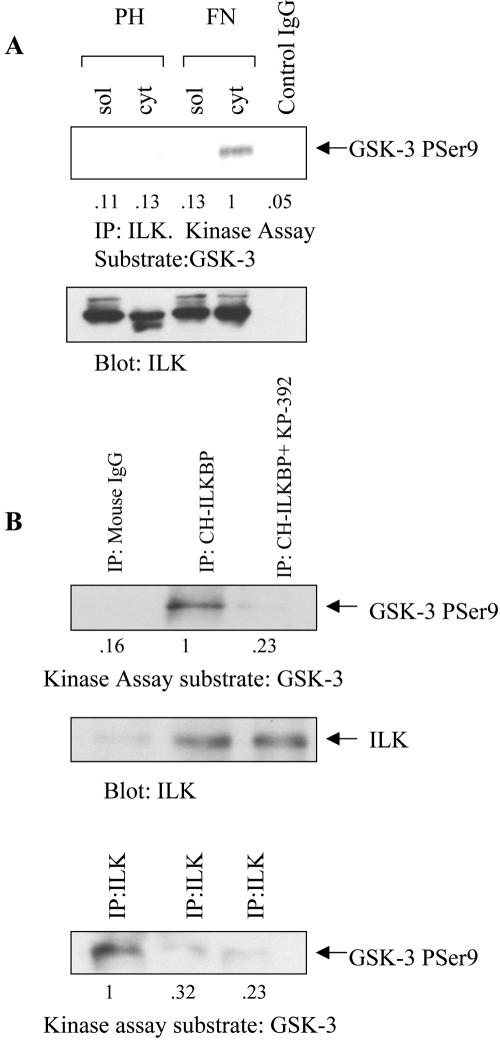
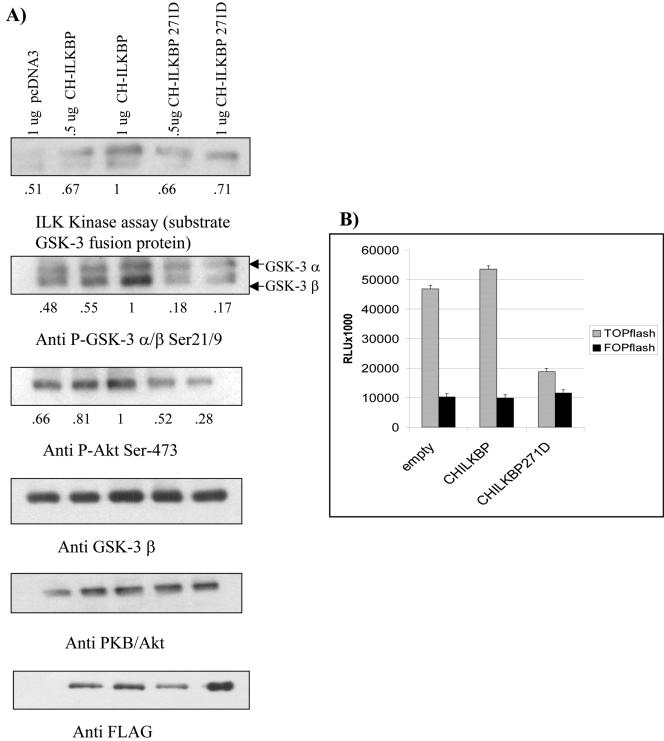
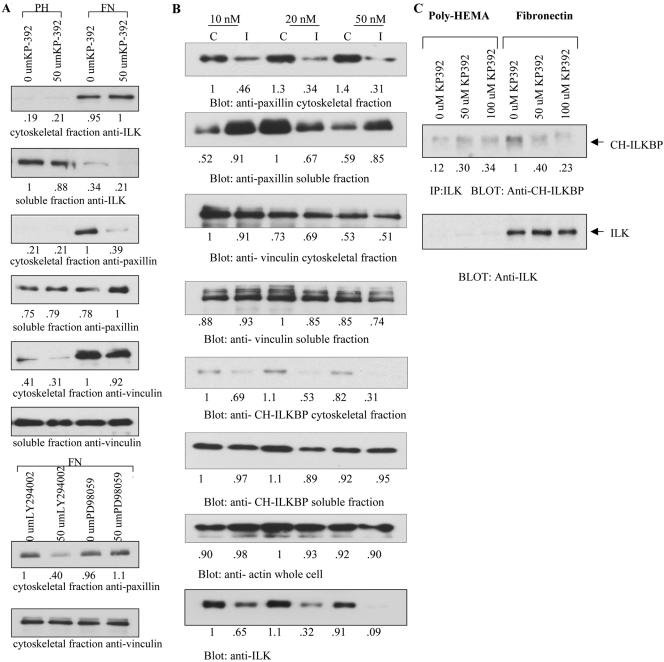
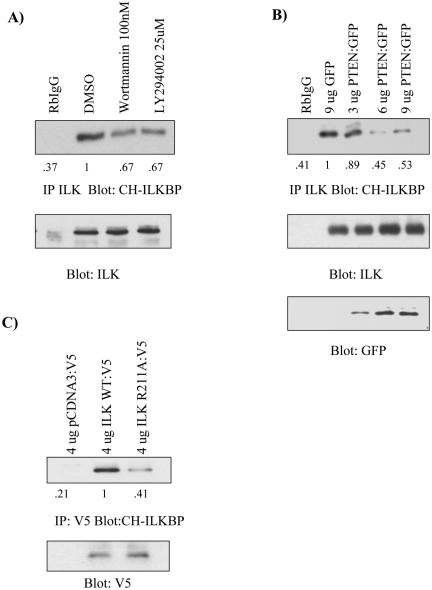
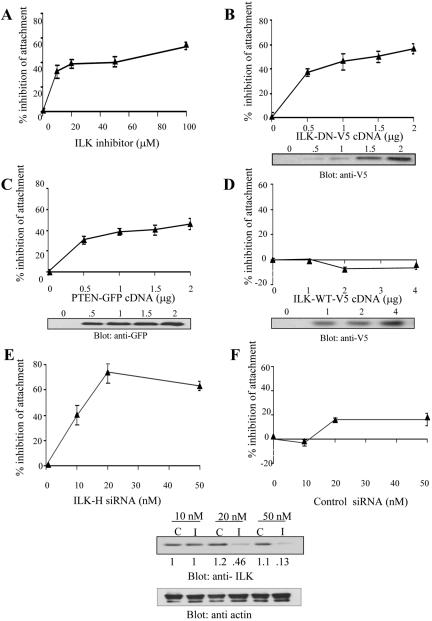
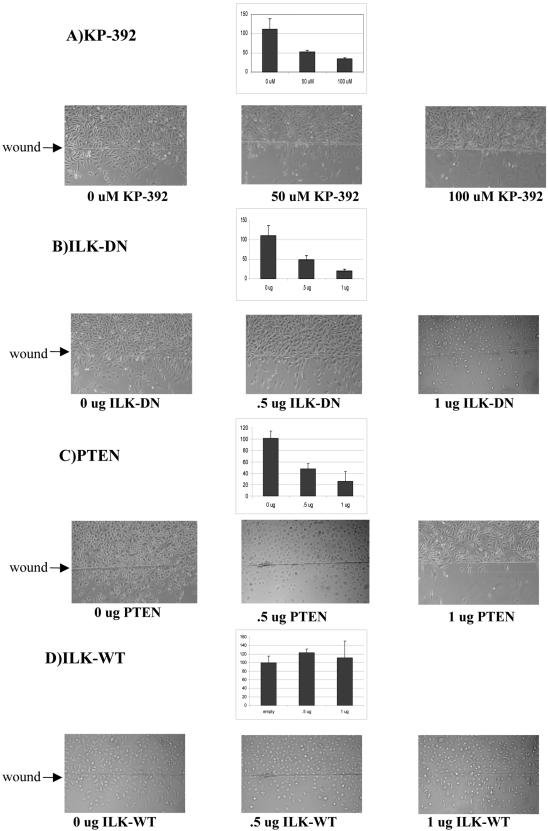
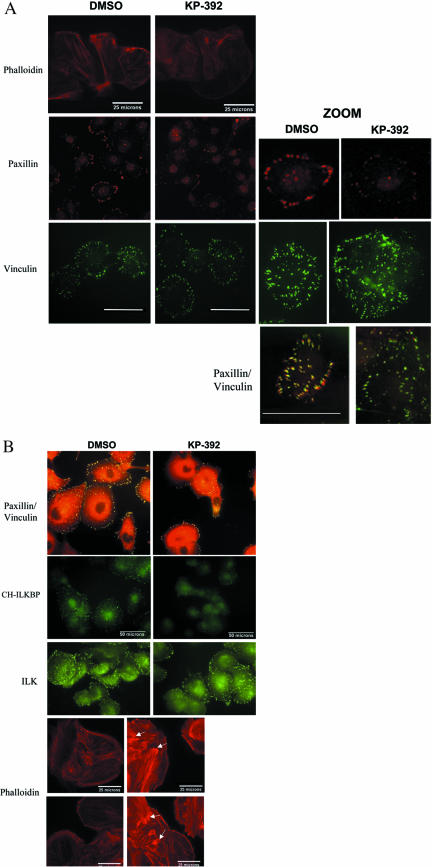
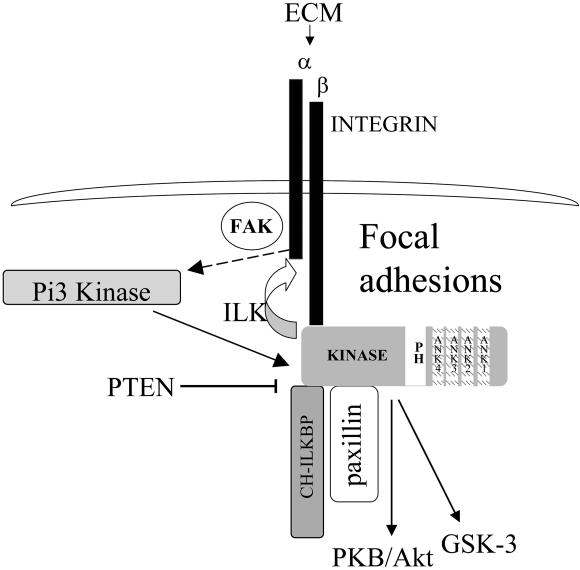
Cell attachment and the assembly of cytoskeletal and signaling complexes downstream of integrins are intimately linked and coordinated. Although many intracellular proteins have been implicated in these processes, a new paradigm is emerging from biochemical and genetic studies that implicates integrin-linked kinase (ILK) and its interacting proteins, such as CH-ILKBP (alpha-parvin), paxillin, and PINCH in coupling integrins to the actin cytoskeleton and signaling complexes. Genetic studies in Drosophila, Caenorhabditis elegans, and mice point to an essential role of ILK as an adaptor protein in mediating integrin-dependent cell attachment and cytoskeletal organization. Here we demonstrate, using several different approaches, that inhibiting ILK kinase activity, or expression, results in the inhibition of cell attachment, cell migration, F-actin organization, and the specific cytoskeletal localization of CH-ILKBP and paxillin in human cells. We also demonstrate that the kinase activity of ILK is elevated in the cytoskeletal fraction and that the interaction of CH-ILKBP with ILK within the cytoskeleton stimulates ILK activity and downstream signaling to PKB/Akt and GSK-3. Interestingly, the interaction of CH-ILKBP with ILK is regulated by the Pi3 kinase pathway, because inhibition of Pi3 kinase activity by pharmacological inhibitors, or by the tumor suppressor PTEN, inhibits this interaction as well as cell attachment and signaling. These data demonstrate that the kinase and adaptor properties of ILK function together, in a Pi3 kinase-dependent manner, to regulate integrin-mediated cell attachment and signal transduction.
Figures
References
-
- Attwell, S., Roskelley, C., and Dedhar, S. (2000). The integrin-liked kinase (ILK) suppresses anoikis. Oncogene 19, 3811–3815. - PubMed
-
- Calderwood, D.A., Shattil, S.J., and Ginsberg, M.H. (2000). Integrins and actin filaments: reciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 275, 22607–22610. - PubMed
-
- Carrieras, F., Rigot, V., Cruet, S., Andre, F., Gauduchon, P., and Marvaldi, J. (1999). Migration properties of the human ovarian adenocarcinoma cell line IGROV 1, Importance of αvβ3 integrins and vitronectin. Int. J. Cancer. 80, 285–94. - PubMed
-
- Cruet-Hennequart, S., Maubant, S., Luis, J., Gauduchon, P., Staedel, C., and Dedhar, S. (2003). αv integrins regulate cell proliferation through integrin-linked kinase (ILK) in ovarian cancer cells. Oncogene 22, 1688–1702. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
