

Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells
- PMID: 12972646
- PMCID: PMC208776
- DOI: 10.1073/pnas.1534570100
Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells
Abstract
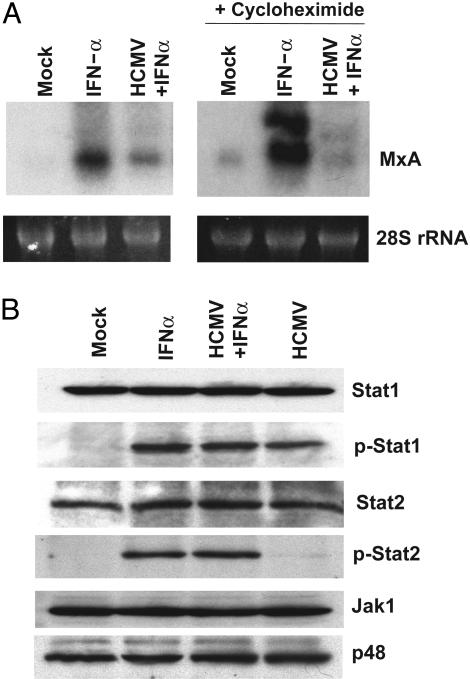
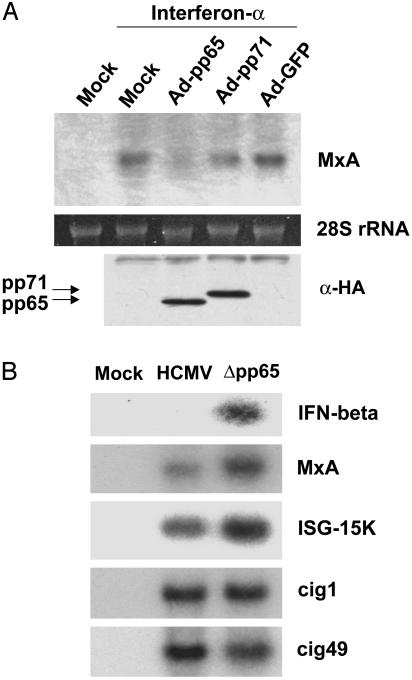
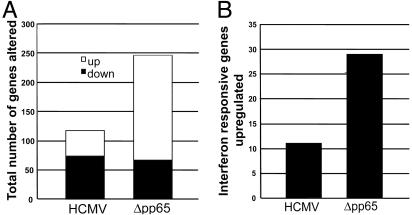
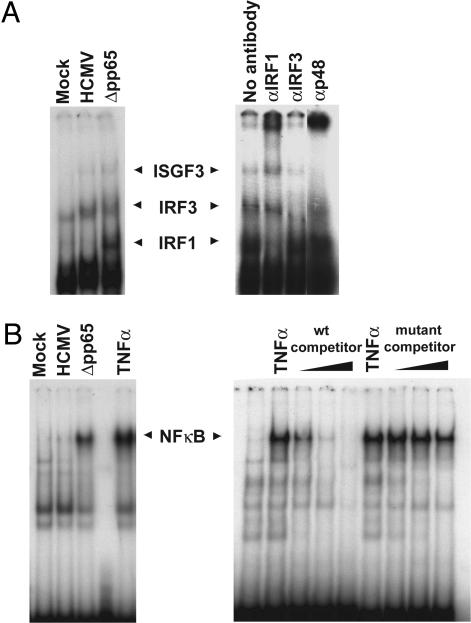
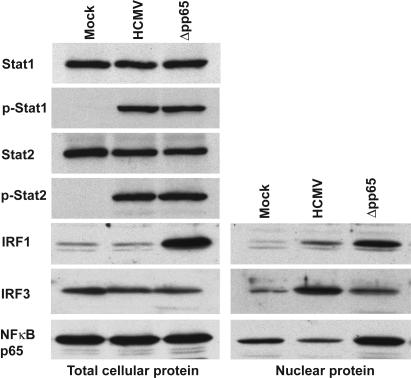
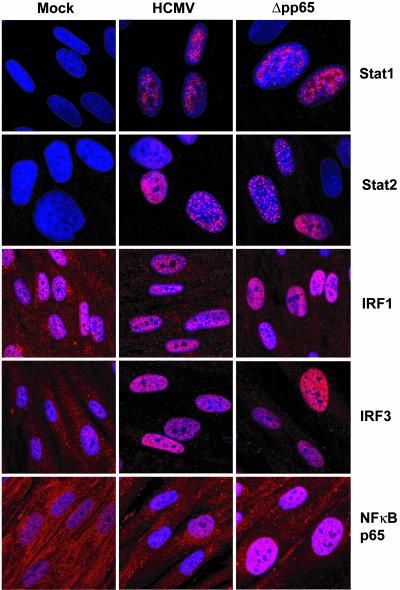
The initial interaction of human cytomegalovirus with fibroblasts triggers, and then partially blocks, an innate immune response pathway that leads to the induction of IFN-responsive genes and proinflammatory chemokines. Infection of fibroblasts with human cytomegalovirus inhibited their ability to respond to exogenous IFN. Consistent with the observation that the block did not depend on de novo viral protein synthesis, ectopic expression of the viral UL83-coded pp65, an abundant virion protein, inhibited IFN signaling. Furthermore, DNA array analysis showed that infection with a pp65-deficient mutant virus caused a much stronger induction of many IFN-response and proinflammatory chemokine RNAs than infection with wild-type virus. The nuclear DNA-binding activities of transcription factors NF-kappaB and IRF1 were induced to a much greater extent after infection with the pp65-deficient mutant than with wild-type virus. IFN-stimulated gene factor 3 DNA-binding was modestly enhanced, whereas IRF3 activity was not affected by mutation of pp65. Together, these results imply that pp65, which is delivered to newly infected cells in the virion, antagonizes a pathway that affects NF-kappaB and IRF1 and prevents the accumulation of mRNAs encoded by numerous cellular antiviral genes.
Figures
References
-
- Mendelson, M., Monard, S., Sissons, P. & Sinclair, J. (1996) J. Gen. Virol. 77, 3099–3102. - PubMed
-
- Jarvis, M. A. & Nelson, J. A. (2002) Curr. Opin. Microbiol. 5, 403–407. - PubMed
-
- Gutermann, A., Bubeck, A., Wagner, M., Reusch, U., Menard, C. & Koszinowski, U. H. (2002) Curr. Top. Microbiol. Immunol. 269, 1–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
