

Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis
- PMID: 12973667
- PMCID: PMC1180616
- DOI: 10.1086/378781
Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis
Abstract
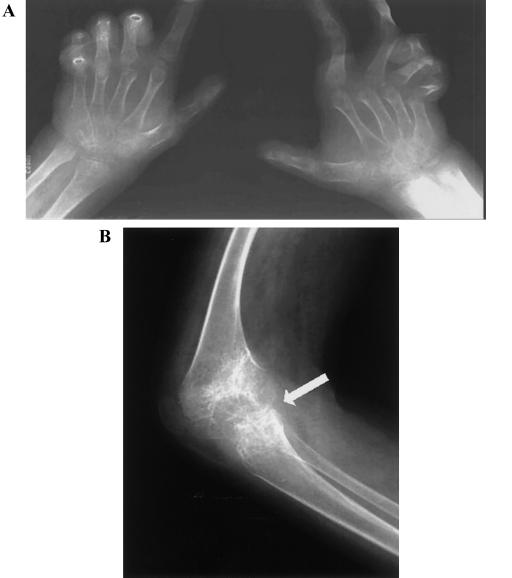
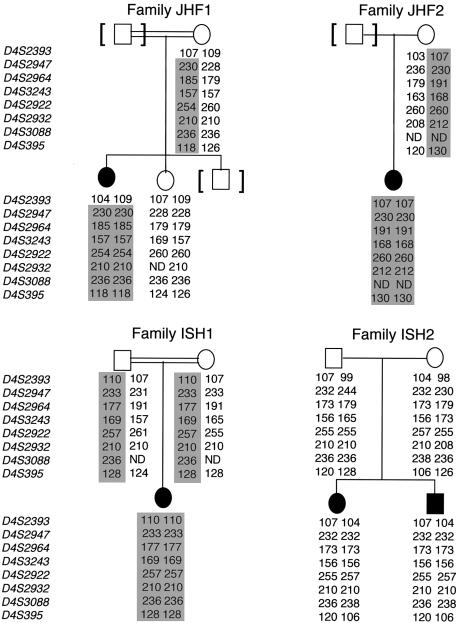
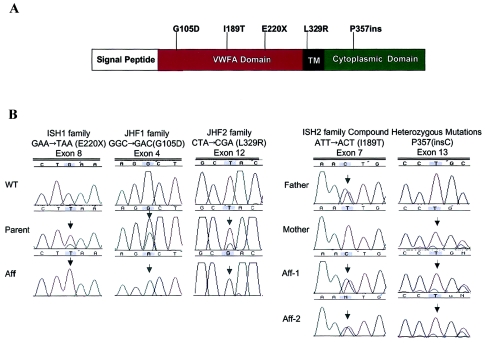
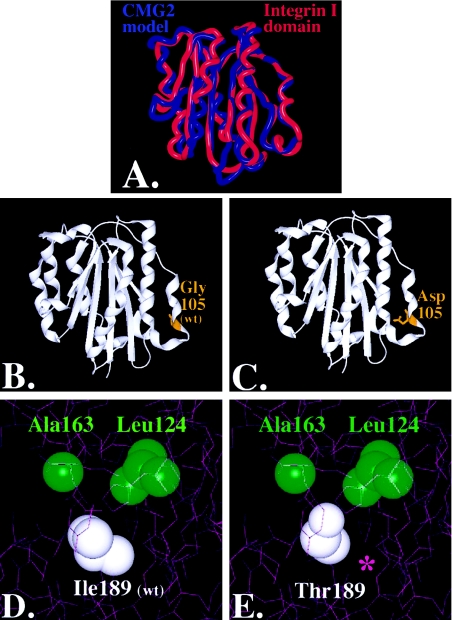
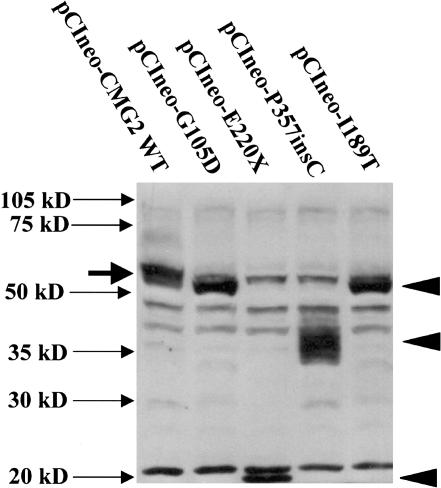
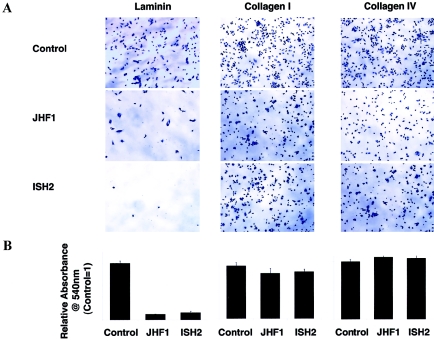
Juvenile hyaline fibromatosis (JHF) and infantile systemic hyalinosis (ISH) are autosomal recessive syndromes of unknown etiology characterized by multiple, recurring subcutaneous tumors, gingival hypertrophy, joint contractures, osteolysis, and osteoporosis. Both are believed to be allelic disorders; ISH is distinguished from JHF by its more severe phenotype, which includes hyaline deposits in multiple organs, recurrent infections, and death within the first 2 years of life. Using the previously reported chromosome 4q21 JHF disease locus as a guide for candidate-gene identification, we identified and characterized JHF and ISH disease-causing mutations in the capillary morphogenesis factor-2 gene (CMG2). Although CMG2 encodes a protein upregulated in endothelial cells during capillary formation and was recently shown to function as an anthrax-toxin receptor, its physiologic role is unclear. Two ISH family-specific truncating mutations, E220X and the 1-bp insertion P357insC that results in translation of an out-of-frame stop codon, were generated by site-directed mutagenesis and were shown to delete the CMG-2 transmembrane and/or cytosolic domains, respectively. An ISH compound mutation, I189T, is predicted to create a novel and destabilizing internal cavity within the protein. The JHF family-specific homoallelic missense mutation G105D destabilizes a von Willebrand factor A extracellular domain alpha-helix, whereas the other mutation, L329R, occurs within the transmembrane domain of the protein. Finally, and possibly providing insight into the pathophysiology of these diseases, analysis of fibroblasts derived from patients with JHF or ISH suggests that CMG2 mutations abrogate normal cell interactions with the extracellular matrix.
Figures
References
Electronic-Database Information
-
- Celera, http://www.celera.com/ (for identification of candidate genes)
-
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/
-
- Decode, http://www.decodegenetics.com/ (for the human genetic map)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for JHF, ISH, epidermolysis bullosa letalis, epidermolysis bullosa with pyloric atresia, and multiple epiphyseal dysplasia)
-
- Protein Data Bank, http://www.rcsb.org/pdb/
References
-
- Bell SE, Mavila A, Salazar R, Bayless KJ, Kanagala S, Maxwell SA, Davis GE (2001) Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J Cell Sci 114:2755–2773 - PubMed
-
- Chapman KL, Mortier GR, Chapman K, Loughlin J, Grant ME, Briggs MD (2001) Mutations in the region encoding the von Willebrand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nat Genet 28:393–396 - PubMed
-
- Colognato H, Yurchenco PD (2000) Form and function: the laminin family of heterotrimers. Dev Dyn 218:213–234 - PubMed
-
- Daluiski A, Engstrand T, Bahamonde ME, Gamer LW, Agius E, Stevenson SL, Cox K, Rosen V, Lyons KM (2001) Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet 27:84–88 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
