

Mutations of the beta myosin heavy chain gene in hypertrophic cardiomyopathy: critical functional sites determine prognosis
- PMID: 12975413
- PMCID: PMC1767874
- DOI: 10.1136/heart.89.10.1179
Mutations of the beta myosin heavy chain gene in hypertrophic cardiomyopathy: critical functional sites determine prognosis
Abstract
Objectives: To assess patients with different types of mutations of the beta myosin heavy chain (beta MHC) gene causing hypertrophic cardiomyopathy (HCM) and to determine the prognosis of patients according to the affected functional domain of beta MHC.
Design and setting: Cohort study of subjects referred to an HCM clinic at an academic hospital.
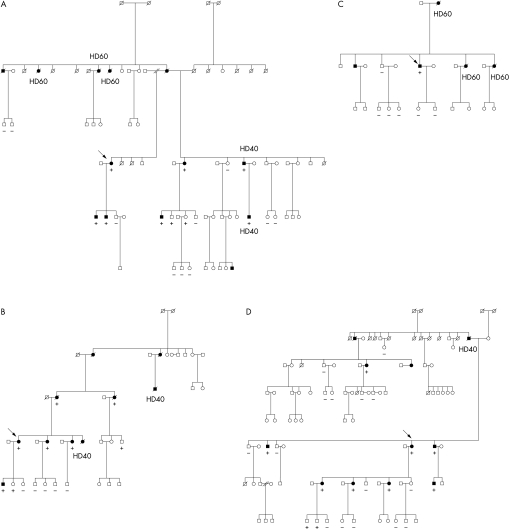
Patients: 70 probands from the HCM clinic were screened for mutations of the beta MHC gene and 148 family members of the genotype positive probands were further assessed. The control group for the genetic studies consisted of 106 healthy subjects.
Main outcome measures: Direct DNA sequencing was used to screen 70 probands for mutations of the beta MHC gene. Family members underwent genotypic and detailed clinical, ECG, and echocardiographic assessments. The survival of genotype positive subjects was evaluated according to the type of functional domain affected by the missense mutation and according to phenotypic characteristics.
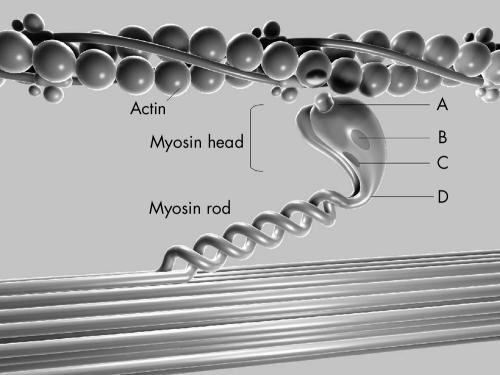
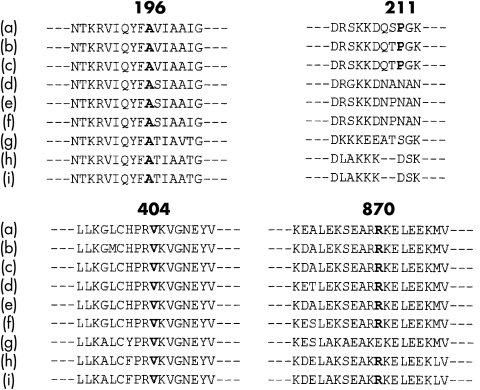
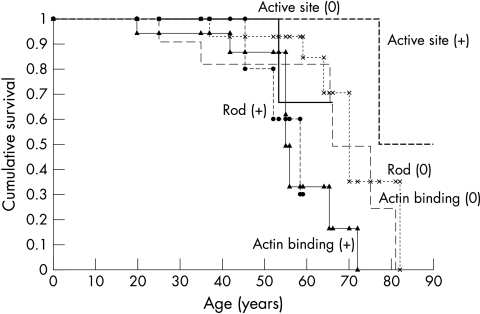
Results: A mutation of the beta MHC gene was detected in 15 of 70 probands (21%). Of 148 family members studied in these 15 families, 74 were identified with a beta MHC defect. Eleven mutations were detected, including four novel mutations: Ala196Thr, Pro211Leu, Val404Leu, and Arg870Cys. Median survival was 66 years (95% confidence interval (CI) 64 to 77 years) in all affected subjects. There was a significant difference in survival between subjects according to the affected functional domain (p = 0.02). Significant independent predictors of decreased survival were the non-conservative (that is, associated with a change in the amino acid charge) missense mutations that affected the actin binding site (hazard ratio 4.4, 95% CI 1.6 to 11.8; p = 0.003) and those that affected the rod portion of beta MHC (hazard ratio 4.8, 95% CI 1.2 to 19.4; p = 0.03). No phenotypic characteristics were associated with decreased survival or cardiovascular morbidity.
Conclusions: The type of beta MHC functional domain affected by the missense mutation is predictive of overall prognosis in HCM.
Figures
Comment in
-
From malignant mutations to malignant domains: the continuing search for prognostic significance in the mutant genes causing hypertrophic cardiomyopathy.Heart. 2004 Jan;90(1):7-8. doi: 10.1136/heart.90.1.7. Heart. 2004. PMID: 14676227 Free PMC article.
Similar articles
-
Clinical and prognostic evaluation of familial hypertrophic cardiomyopathy in two South African families with different cardiac beta myosin heavy chain gene mutations.Br Heart J. 1995 Jul;74(1):40-6. doi: 10.1136/hrt.74.1.40. Br Heart J. 1995. PMID: 7662452 Free PMC article.
-
Coexistence of mitochondrial DNA and beta myosin heavy chain mutations in hypertrophic cardiomyopathy with late congestive heart failure.Heart. 1998 Dec;80(6):548-58. doi: 10.1136/hrt.80.6.548. Heart. 1998. PMID: 10065021 Free PMC article.
-
Sudden cardiac death in hypertrophic cardiomyopathy. Variability in phenotypic expression of beta-myosin heavy chain mutations.Eur Heart J. 1995 Mar;16(3):368-76. doi: 10.1093/oxfordjournals.eurheartj.a060920. Eur Heart J. 1995. PMID: 7789380
-
[Genetic changes and clinical management in familial hypertrophic cardiomyopathy].Wiad Lek. 2000;53(1-2):4-21. Wiad Lek. 2000. PMID: 10806915 Review. Polish.
-
Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives.J Am Coll Cardiol. 2012 Aug 21;60(8):705-15. doi: 10.1016/j.jacc.2012.02.068. Epub 2012 Jul 11. J Am Coll Cardiol. 2012. PMID: 22796258 Review.
Cited by
-
Phenotype and prognostic correlations of the converter region mutations affecting the β myosin heavy chain.Heart. 2015 Jul;101(13):1047-53. doi: 10.1136/heartjnl-2014-307205. Epub 2015 May 2. Heart. 2015. PMID: 25935763 Free PMC article.
-
Arrhythmogenic Cardiomyopathy: Exercise Pitfalls, Role of Connexin-43, and Moving beyond Antiarrhythmics.Int J Mol Sci. 2022 Aug 6;23(15):8753. doi: 10.3390/ijms23158753. Int J Mol Sci. 2022. PMID: 35955883 Free PMC article. Review.
-
Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy.Circulation. 2010 Dec 7;122(23):2430-40; discussion 2440. doi: 10.1161/CIRCULATIONAHA.110.978924. Circulation. 2010. PMID: 21135371 Free PMC article. Review. No abstract available.
-
Successful pregnancy and delivery in a young-onset hypertrophic cardiomyopathy patient with a novel doublet-base substitution in the MYH7 gene.J Cardiol Cases. 2022 Oct 1;27(1):8-11. doi: 10.1016/j.jccase.2022.09.010. eCollection 2023 Jan. J Cardiol Cases. 2022. PMID: 36618848 Free PMC article.
-
Recessive myosin myopathy with external ophthalmoplegia associated with MYH2 mutations.Eur J Hum Genet. 2014 Jun;22(6):801-8. doi: 10.1038/ejhg.2013.250. Epub 2013 Nov 6. Eur J Hum Genet. 2014. PMID: 24193343 Free PMC article.
References
-
- Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 2001;104:557–67. - PubMed
-
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002;287:1308–20. - PubMed
-
- Roberts R, Sigwart U. New concepts in hypertrophic cardiomyopathies, part I. Circulation 2001;104:2113–16. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials