

Multiple steps of phosphorylation of activated rhodopsin can account for the reproducibility of vertebrate rod single-photon responses
- PMID: 12975449
- PMCID: PMC1480412
- DOI: 10.1085/jgp.200308832
Multiple steps of phosphorylation of activated rhodopsin can account for the reproducibility of vertebrate rod single-photon responses
Abstract
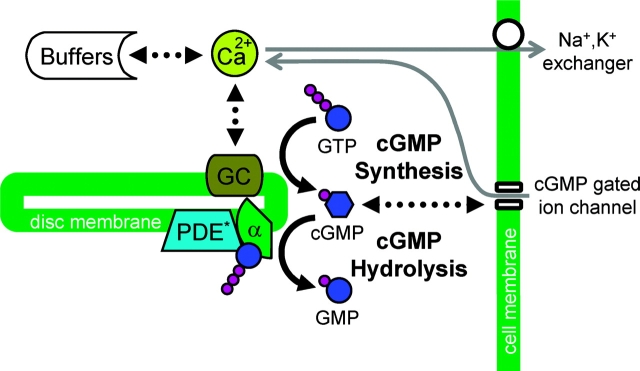
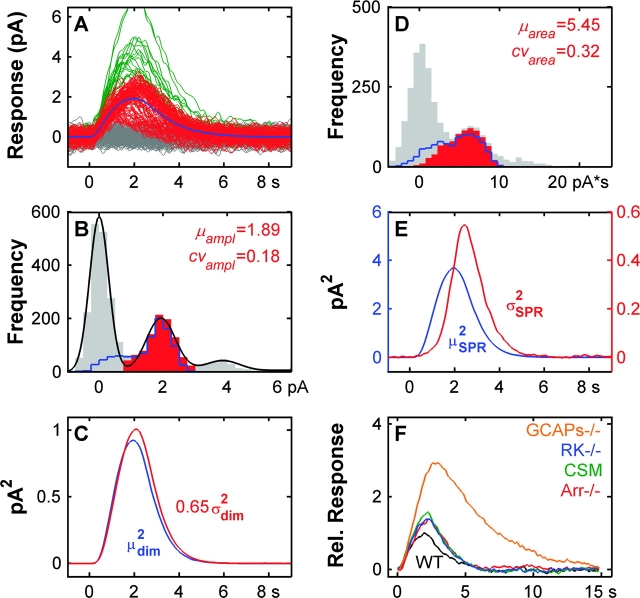
Single-photon responses (SPRs) in vertebrate rods are considerably less variable than expected if isomerized rhodopsin (R*) inactivated in a single, memoryless step, and no other variability-reducing mechanisms were available. We present a new stochastic model, the core of which is the successive ratcheting down of R* activity, and a concomitant increase in the probability of quenching of R* by arrestin (Arr), with each phosphorylation of R* (Gibson, S.K., J.H. Parkes, and P.A. Liebman. 2000. Biochemistry. 39:5738-5749.). We evaluated the model by means of Monte-Carlo simulations of dim-flash responses, and compared the response statistics derived from them with those obtained from empirical dim-flash data (Whitlock, G.G., and T.D. Lamb. 1999. Neuron. 23:337-351.). The model accounts for four quantitative measures of SPR reproducibility. It also reproduces qualitative features of rod responses obtained with altered nucleotide levels, and thus contradicts the conclusion that such responses imply that phosphorylation cannot dominate R* inactivation (Rieke, F., and D.A. Baylor. 1998a. Biophys. J. 75:1836-1857; Field, G.D., and F. Rieke. 2002. Neuron. 35:733-747.). Moreover, the model is able to reproduce the salient qualitative features of SPRs obtained from mouse rods that had been genetically modified with specific pathways of R* inactivation or Ca2+ feedback disabled. We present a theoretical analysis showing that the variability of the area under the SPR estimates the variability of integrated R* activity, and can provide a valid gauge of the number of R* inactivation steps. We show that there is a heretofore unappreciated tradeoff between variability of SPR amplitude and SPR duration that depends critically on the kinetics of inactivation of R* relative to the net kinetics of the downstream reactions in the cascade. Because of this dependence, neither the variability of SPR amplitude nor duration provides a reliable estimate of the underlying variability of integrated R* activity, and cannot be used to estimate the minimum number of R* inactivation steps. We conclude that multiple phosphorylation-dependent decrements in R* activity (with Arr-quench) can confer the observed reproducibility of rod SPRs; there is no compelling need to invoke a long series of non-phosphorylation dependent state changes in R* (as in Rieke, F., and D.A. Baylor. 1998a. Biophys. J. 75:1836-1857; Field, G.D., and F. Rieke. 2002. Neuron. 35:733-747.). Our analyses, plus data and modeling of others (Rieke, F., and D.A. Baylor. 1998a. Biophys. J. 75:1836-1857; Field, G.D., and F. Rieke. 2002. Neuron. 35:733-747.), also argue strongly against either feedback (including Ca2+-feedback) or depletion of any molecular species downstream to R* as the dominant cause of SPR reproducibility.
Figures






References
-
- Arshavsky, V.Y., T.D. Lamb, and E.N. Pugh. 2002. G proteins and phototransduction. Annu. Rev. Physiol. 64:153–187. - PubMed
-
- Aton, B.R., B.J. Litman, and M.L. Jackson. 1984. Isolation and identification of the phosphorylated species of rhodopsin. Biochemistry. 23:1737–1741. - PubMed
-
- Burns, M.E., A. Mendez, J. Chen, and D.A. Baylor. 2002. Dynamics of cyclic GMP synthesis in retinal rods. Neuron. 36:81–91. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
