

Review
doi: 10.1172/JCI19844.
Long QT syndrome: novel insights into the mechanisms of cardiac arrhythmias
Affiliations
- PMID: 12975462
- PMCID: PMC193679
- DOI: 10.1172/JCI19844
Item in Clipboard
Review
Long QT syndrome: novel insights into the mechanisms of cardiac arrhythmias
J Clin Invest.
2003 Sep.
Abstract
The congenital long QT syndrome is a rare disorder in which mutation carriers are at risk for polymorphic ventricular tachycardia and/or sudden cardiac death. Discovery and analysis of gene mutations associated with variants of this disorder have provided novel insight into mechanisms of cardiac arrhythmia and have raised the possibility of mutation-specific therapeutic intervention.
Figures
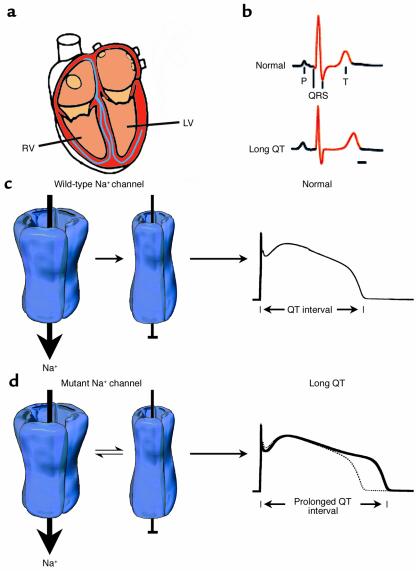
Mutation-altered Na+ channel inactivation underlies the LQT-3 phenotype. (a) Schematic view of the human heart emphasizing right ventricle (RV) and left ventricle (LV), which contain the substrates for altered electrical signaling in LQTS. (b) The clinical phenotype of LQTS is a prolonged QT interval of the ECG. (c) Gating of normal (wild-type) human heart Na+ channels during the action potential plateau. Channels rapidly enter an absorbing inactivated state from which they rarely reopen, and thus the QT interval (far right) is not greatly affected by wild-type Na+ channels. (d) Na+ channel gating in most forms of LQT-3 syndrome is altered. During the plateau phase, channels enter a mode of gating in which inactivation is no longer an absorbing state. A small fraction of channels open, enter a nonconducting inactivated state, and then transition back and forth between open and nonconducting states for the duration of the action potential plateau. This produces a small depolarizing current that prolongs the QT interval of the ECG of mutation carriers (far right). Modified with permission from refs. and .

Disruption of local signaling domains occurs in LQT-1 syndrome. Stimulation of β-adrenergic receptors (β-ARs) in the heart leads to PKA-dependent phosphorylation of multiple intracellular targets in cardiac myocytes. These targets include the ryanodine receptor (RyR2), L-type calcium channels, and the KCNQ1/KCNE1 K+ channel. In response to stress in healthy patients, β-AR stimulation results in phosphorylation of all three of these targets and uniform electrical activity on the ECG (upper ECG). When the KCNQ1/KCNE1 complex is disrupted by an inherited mutation, an unbalanced cellular response occurs, which leads to dysfunctional rhythm (lower ECG). Modified with permission from ref. . SR, sarcoplasmic reticulum; VGCC, voltage-gated calcium channel.
References
-
- Jervell A, Lange-Nielsen F. Congential deafmutism, functional heart disease with prolongation of the Q interval, and sudden death. Am. Heart J. 1957;54:59–68. - PubMed
-
- Levine SA, Woodworth CR. Congenital deaf-mutism, prolonged Q-T interval, syncopal attacks and sudden death. N. Engl. J. Med. 1958;259:412–417. - PubMed
-
- Romano C, Gemme G, Pongiglione R. Artimie cardiache rare dell’eta pediatria. Clin. Pediatr. (Phila.). 1963;45:656–683. - PubMed
-
- Ward OC. New familial cardiac syndrome in children. J. Ir. Med. Assoc. 1964;54:103–106. - PubMed
-
- Moss AJ, Schwartz PJ. Delayed repolarization (QT or QTU prolongation) and malignant ventricular arrhythmias. Mod. Concepts Cardiovasc. Dis. 1982;51:85–90. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
