

Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene
- PMID: 1346338
- PMCID: PMC6151859
- DOI: 10.1056/NEJM199202133260704
Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene
Abstract
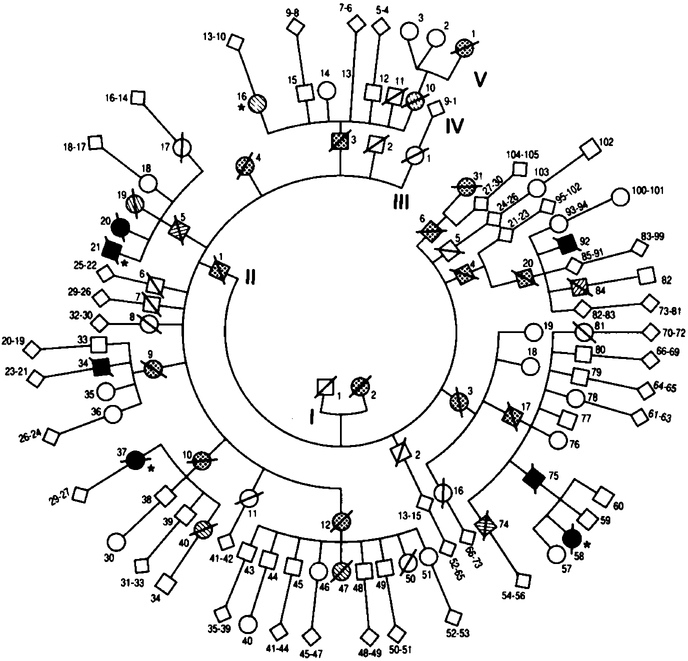
Background: We previously described two members of a family affected by an apparently genetically determined fatal disease characterized clinically by progressive insomnia, dysautonomia, and motor signs and characterized pathologically by severe atrophy of the anterior ventral and mediodorsal thalamic nuclei. Five other family members who died of this disease, which we termed "fatal familial insomnia," had broader neuropathologic changes suggesting that fatal familial insomnia could be a prion disease.
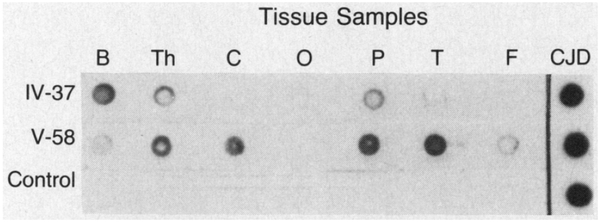
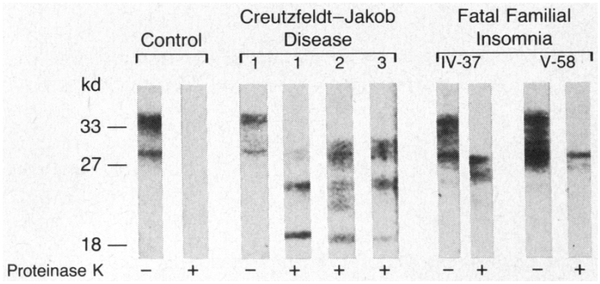
Methods: We used antibodies to prion protein (PrP) to perform dot and Western blot analyses, with and without proteinase K, on brain tissue obtained at autopsy from two patients with fatal familial insomnia, three patients with sporadic Creutzfeldt-Jakob disease, and six control subjects. The coding region of the PrP gene was amplified and sequenced in the samples from the two patients with fatal familial insomnia. Restriction-enzyme analysis was carried out with amplified PrP DNA from 33 members of the kindred.
Results: Protease-resistant PrP was found in both patients with fatal familial insomnia, but the size and number of protease-resistant fragments differed from those in Creutzfeldt-Jakob disease. In the family with fatal familial insomnia, all 4 affected members and 11 of the 29 unaffected members had a point mutation in PrP codon 178 that results in the substitution of asparagine for aspartic acid and elimination of the Tth111 I restriction site. Linkage analysis showed a close relation between the point mutation and the disease (maximal lod score, 3.4 when theta was zero).
Conclusions: Fatal familial insomnia is a prion disease with a mutation in codon 178 of the PrP gene, but the disease phenotype seems to differ from that of previously described kindreds with the same point mutation.
Figures


Comment in
-
Prion disease.N Engl J Med. 1992 Feb 13;326(7):486-7. doi: 10.1056/NEJM199202133260711. N Engl J Med. 1992. PMID: 1346339 No abstract available.
References
-
- Lugaresi E,Medori R,Montagna P, et al. Fatal familialin somnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 1986;315:997–1003. - PubMed
-
- Hirai T, Jones EG. A new parcellation of the human thalamus on the basis of histochemical staining. Brain Res Brain Res Rev 1989;14:1–34. - PubMed
-
- Manetto V, Medori R, Cortelli P, et al. Fatal familialin somnia clinical and pathological study of five new cases. Neurology(in press). - PubMed
-
- Prusiner SB. Molecular biology of prion diseases. Science 1991;252:1515–22. - PubMed
-
- Serban D, Taraboulos A, DeArmond SJ, Prusiner SB. Rapid detection of Creutzfeldt-Jakob disease and scrapie prion proteins. Neurology 1990;40: 110–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials