

Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition
- PMID: 14504389
- PMCID: PMC208801
- DOI: 10.1073/pnas.2034960100
Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition
Abstract
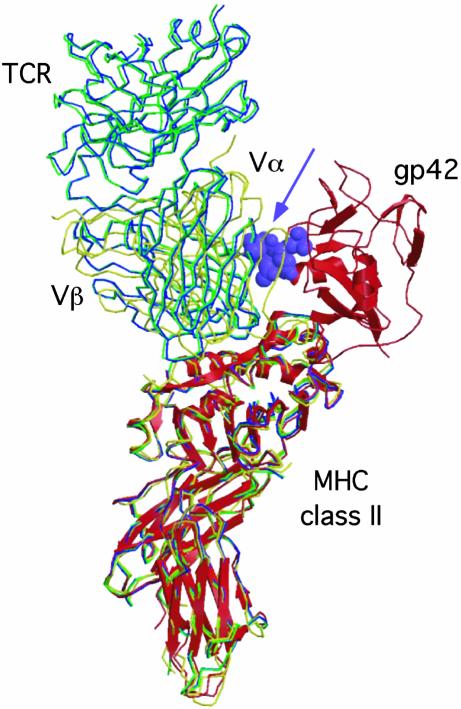
Epstein-Barr virus (EBV) persists lifelong in infected hosts despite the presence of antiviral immunity. Many viral antigens are expressed during lytic infection. Thus, for EBV to spread, it must have evolved effective ways to evade immune recognition. Here, we report that HLA class II-restricted antigen presentation to T helper cells is hampered in the presence of the lytic-phase protein gp42. This interference with T cell activation involves association of gp42 with class II peptide complexes. Using HLA-DR tetramers, we identify a block in T cell receptor (TCR)-class II interactions imposed by gp42 as the underlying mechanism. EBV gp42 sterically clashes with TCR Valpha-domains as visualized by superimposing the crystal structures for gp42-HLA-DR1 and TCR-MHC class II complexes. Blocking TCR recognition provides a previously undescribed strategy for viral immune evasion.
Figures
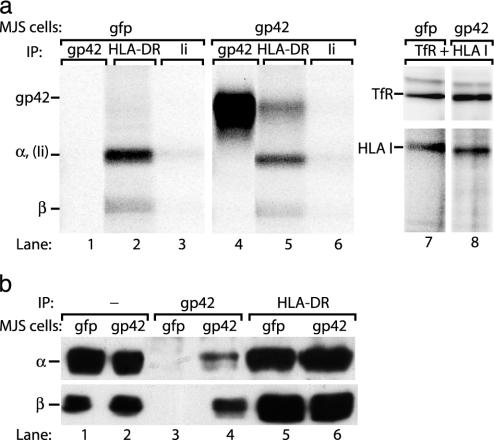
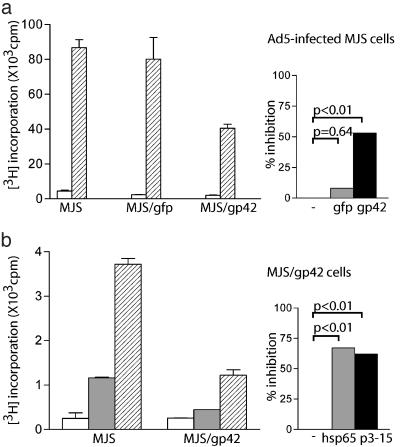
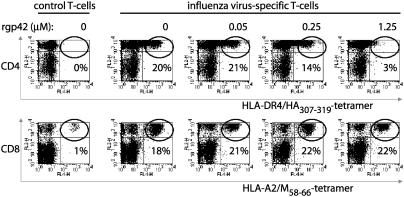
 ) to R30.95 T cells (a) or the M. tuberculosis hsp65 protein (
) to R30.95 T cells (a) or the M. tuberculosis hsp65 protein ( ) and the related p3-15 peptide () to Rp15.1.1 T cells (b). [3H]Thymidine incorporation is depicted with error bars for triplicates. (Right) Percentage inhibition of antigen-specific T cell proliferation in response to MJS/gp42 or MJS/gfp cells is depicted compared with (uninfected) MJS cells. Statistical analysis on triplicate wells was performed by the Student's t test, and P values are indicated.
) and the related p3-15 peptide () to Rp15.1.1 T cells (b). [3H]Thymidine incorporation is depicted with error bars for triplicates. (Right) Percentage inhibition of antigen-specific T cell proliferation in response to MJS/gp42 or MJS/gfp cells is depicted compared with (uninfected) MJS cells. Statistical analysis on triplicate wells was performed by the Student's t test, and P values are indicated.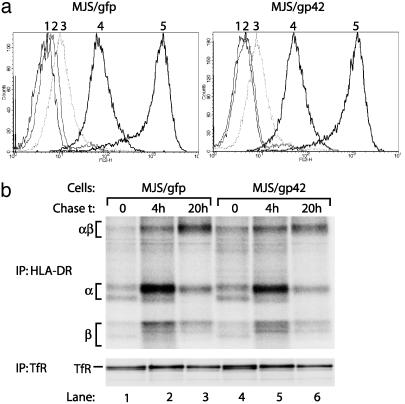
References
-
- Tortorella, D., Gewurz, B. E., Furman, M. H., Schust, D. J. & Ploegh, H. L. (2000) Annu. Rev. Immunol. 18, 861–926. - PubMed
-
- Cresswell, P. (1994) Annu. Rev. Immunol. 12, 259–293. - PubMed
-
- Rickinson, A. B. & Kieff, E. (2001) in Field's Virology, eds. Knipe, D. M. & Howley, P. M. (Lippincott Williams & Wilkins, Philadelphia), pp. 2575–2627.
-
- Rickinson, A. B. & Moss, D. J. (1997) Annu. Rev. Immunol. 15, 405–431. - PubMed
-
- Levitsky, V. & Masucci, M. G. (2002) Virus Res. 88, 71–86. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
