

NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6)
- PMID: 14508710
- PMCID: PMC1180617
- DOI: 10.1086/378817
NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6)
Abstract
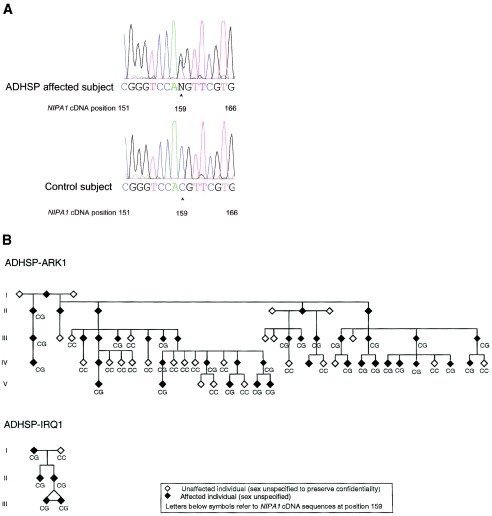
The hereditary spastic paraplegias (HSPs) are genetically heterogeneous disorders characterized by progressive lower-extremity weakness and spasticity. The molecular pathogenesis is poorly understood. We report discovery of a dominant negative mutation in the NIPA1 gene in a kindred with autosomal dominant HSP (ADHSP), linked to chromosome 15q11-q13 (SPG6 locus); and precisely the same mutation in an unrelated kindred with ADHSP that was too small for meaningful linkage analysis. NIPA1 is highly expressed in neuronal tissues and encodes a putative membrane transporter or receptor. Identification of the NIPA1 function and ligand will aid an understanding of axonal neurodegeneration in HSP and may have important therapeutic implications.
Figures

Similar articles
-
A novel NIPA1 mutation associated with a pure form of autosomal dominant hereditary spastic paraplegia.Neurogenetics. 2005 May;6(2):79-84. doi: 10.1007/s10048-004-0209-9. Epub 2005 Feb 12. Neurogenetics. 2005. PMID: 15711826
-
NIPA1 mutation in complex hereditary spastic paraplegia with epilepsy.Eur J Neurol. 2011 Sep;18(9):1197-9. doi: 10.1111/j.1468-1331.2011.03359.x. Epub 2011 Feb 22. Eur J Neurol. 2011. PMID: 21599812
-
Clinical and genetic study of a Brazilian family with spastic paraplegia (SPG6 locus).Mov Disord. 2006 Feb;21(2):279-81. doi: 10.1002/mds.20775. Mov Disord. 2006. PMID: 16267846
-
Clinical and genetic characterization of NIPA1 mutations in a Taiwanese cohort with hereditary spastic paraplegia.Ann Clin Transl Neurol. 2023 Mar;10(3):353-362. doi: 10.1002/acn3.51724. Epub 2023 Jan 6. Ann Clin Transl Neurol. 2023. PMID: 36607129 Free PMC article. Review.
-
SPG6 (NIPA1 variant): A report of a case with early-onset complex hereditary spastic paraplegia and brief literature review.J Clin Neurosci. 2021 Dec;94:281-285. doi: 10.1016/j.jocn.2021.10.026. Epub 2021 Nov 9. J Clin Neurosci. 2021. PMID: 34863451 Review.
Cited by
-
Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlations.J Med Genet. 2006 Jun;43(6):512-6. doi: 10.1136/jmg.2005.036913. Epub 2005 Sep 23. J Med Genet. 2006. PMID: 16183798 Free PMC article.
-
Genetic mutation analysis of hereditary spastic paraplegia: A retrospective study.Medicine (Baltimore). 2020 Jun 5;99(23):e20193. doi: 10.1097/MD.0000000000020193. Medicine (Baltimore). 2020. PMID: 32501971 Free PMC article.
-
Cellular magnesium homeostasis.Arch Biochem Biophys. 2011 Aug 1;512(1):1-23. doi: 10.1016/j.abb.2011.05.010. Epub 2011 May 27. Arch Biochem Biophys. 2011. PMID: 21640700 Free PMC article. Review.
-
Truncating mutation in intracellular phospholipase A₁ gene (DDHD2) in hereditary spastic paraplegia with intellectual disability (SPG54).BMC Res Notes. 2015 Jun 27;8:271. doi: 10.1186/s13104-015-1227-4. BMC Res Notes. 2015. PMID: 26113134 Free PMC article.
-
Phenotypic variability and genetic susceptibility to genomic disorders.Hum Mol Genet. 2010 Oct 15;19(R2):R176-87. doi: 10.1093/hmg/ddq366. Epub 2010 Aug 31. Hum Mol Genet. 2010. PMID: 20807775 Free PMC article. Review.
References
Electronic-Database Information
-
- National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/entrez/query.fcgi
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
-
- Chai J-H, Locke DP, Greally JM, Knoll JHM, Ohta T, Dunai J, Yavor A, Eichler EE, Nicholls R D (2003) Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet 73:898–925 (in this issue) - PMC - PubMed
-
- Fink JK (2001) Hereditary spastic paraplegia. In: Rimoin DPRCJ, Emery KB (eds) Emery & Rimoin's principles and practice of medical genetics. Harcourt Publishers Limited UK, London, pp 3124–3145
-
- Fink JK (2002) Hereditary spastic paraplegia: the pace quickens. Ann Neurol 51:669–672 - PubMed
-
- Fink JK, Hedera P (1999) Hereditary spastic paraplegia: genetic heterogeneity and genotype-phenotype correlation. Semin Neurol 19:301–310 - PubMed
-
- Fink JK, Sharp G, Lange B, Wu C-TB, Haley T, Otterud B, Peacock M, Leppert M (1995a) Autosomal dominant hereditary spastic paraparesis, type I: clinical and genetic analysis of a large North American family. Neurology 45:325–331 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
