

Aichi virus leader protein is involved in viral RNA replication and encapsidation
- PMID: 14512530
- PMCID: PMC224959
- DOI: 10.1128/jvi.77.20.10799-10807.2003
Aichi virus leader protein is involved in viral RNA replication and encapsidation
Abstract
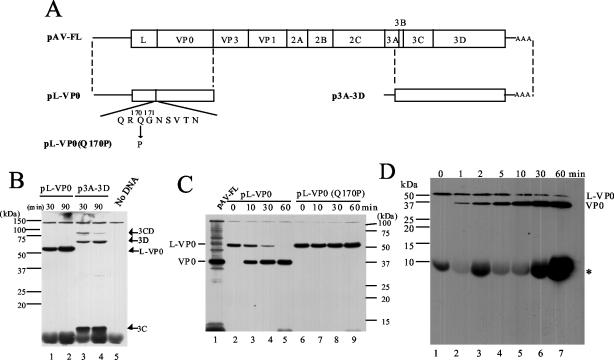
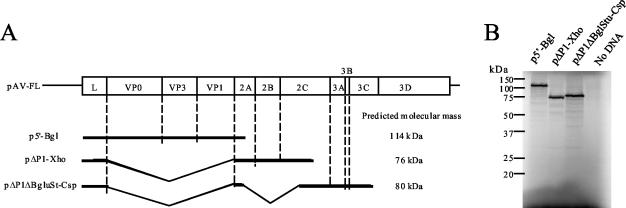
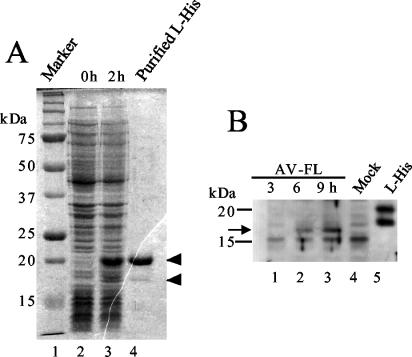
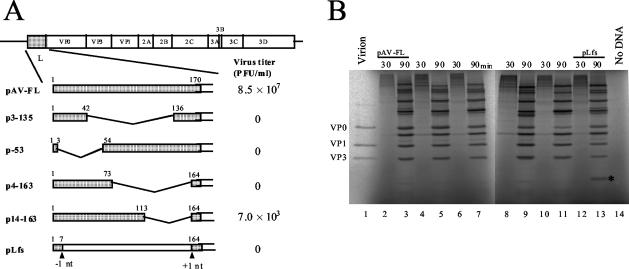
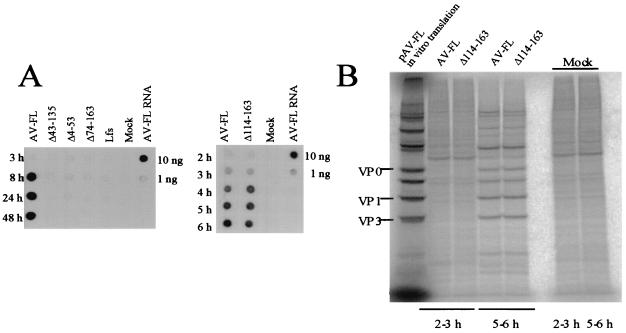
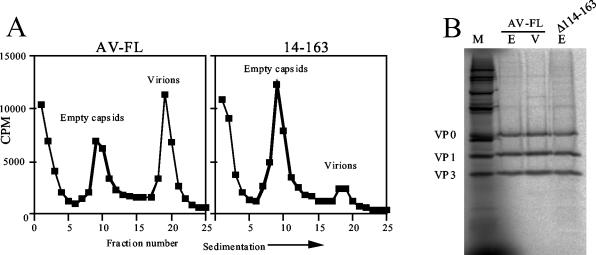
Aichi virus, a member of the family Picornaviridae, encodes a leader (L) protein of 170 amino acids (aa). The Aichi virus L protein exhibits no significant sequence homology to those of other picornaviruses. In this study, we investigated the function of the Aichi virus L protein in virus growth. In vitro translation and cleavage assays indicated that the L protein has no autocatalytic activity and is not involved in polyprotein cleavage. The L-VP0 junction was cleaved by 3C proteinase. Immunoblot analysis showed that the L protein is stably present in infected cells. Characterization of various L mutants derived from an infectious cDNA clone revealed that deletion of 93 aa of the center part (aa 43 to 135), 50 aa of the N-terminal part (aa 4 to 53), or 90 aa of the C-terminal part (aa 74 to 163) abolished viral RNA replication. A mutant (Delta114-163) in which 50 aa of the C-terminal part (aa 114 to 163) were deleted exhibited efficient RNA replication and translation abilities, but the virus yield was 4 log orders lower than that of the wild type. Sedimentation analysis of viral particles generated in mutant Delta114-163 RNA-transfected cells showed that the mutant has a severe defect in the formation of mature virions, but not in that of empty capsids. Thus, the data obtained in this study indicate that the Aichi virus L protein is involved in both viral RNA replication and encapsidation.
Figures
Similar articles
-
Construction of an infectious cDNA clone of Aichi virus (a new member of the family Picornaviridae) and mutational analysis of a stem-loop structure at the 5' end of the genome.J Virol. 2001 Sep;75(17):8021-30. doi: 10.1128/jvi.75.17.8021-8030.2001. J Virol. 2001. PMID: 11483747 Free PMC article.
-
The 5'-end sequence of the genome of Aichi virus, a picornavirus, contains an element critical for viral RNA encapsidation.J Virol. 2003 Mar;77(6):3542-8. doi: 10.1128/jvi.77.6.3542-3548.2003. J Virol. 2003. PMID: 12610129 Free PMC article.
-
The 5'-terminal region of the Aichi virus genome encodes cis-acting replication elements required for positive- and negative-strand RNA synthesis.J Virol. 2005 Jun;79(11):6918-31. doi: 10.1128/JVI.79.11.6918-6931.2005. J Virol. 2005. PMID: 15890931 Free PMC article.
-
[Analysis of Aichi virus replication].Uirusu. 2007 Jun;57(1):67-74. doi: 10.2222/jsv.57.67. Uirusu. 2007. PMID: 18040156 Review. Japanese.
-
Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells.Viruses. 2016 Mar 17;8(3):82. doi: 10.3390/v8030082. Viruses. 2016. PMID: 26999188 Free PMC article. Review.
Cited by
-
The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication.J Virol. 2005 Nov;79(21):13399-411. doi: 10.1128/JVI.79.21.13399-13411.2005. J Virol. 2005. PMID: 16227261 Free PMC article.
-
Isolation and molecular characterization of Aichi viruses from fecal specimens collected in Japan, Bangladesh, Thailand, and Vietnam.J Clin Microbiol. 2007 Jul;45(7):2287-8. doi: 10.1128/JCM.00525-07. Epub 2007 May 23. J Clin Microbiol. 2007. PMID: 17522267 Free PMC article.
-
Novel picornavirus in domestic rabbits (Oryctolagus cuniculus var. domestica).Infect Genet Evol. 2016 Jan;37:117-22. doi: 10.1016/j.meegid.2015.11.012. Epub 2015 Nov 15. Infect Genet Evol. 2016. PMID: 26588888 Free PMC article.
-
The Host Protein CAD Regulates the Replication of FMDV through the Function of Pyrimidines' De Novo Synthesis.J Virol. 2023 May 31;97(5):e0036923. doi: 10.1128/jvi.00369-23. Epub 2023 May 10. J Virol. 2023. PMID: 37162335 Free PMC article.
-
Epidemiology of human and animal kobuviruses.Virusdisease. 2014;25(2):195-200. doi: 10.1007/s13337-014-0200-5. Epub 2014 Feb 26. Virusdisease. 2014. PMID: 25674585 Free PMC article.
References
-
- Bienz, K., D. Egger, and L. Pasamontes. 1987. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 160:220-226. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
