

c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling
- PMID: 14517246
- PMCID: PMC204458
- DOI: 10.1093/emboj/cdg474
c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling
Abstract
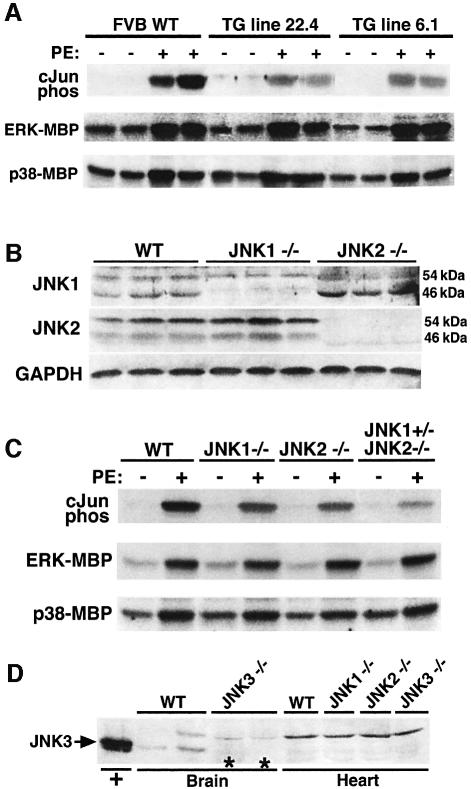
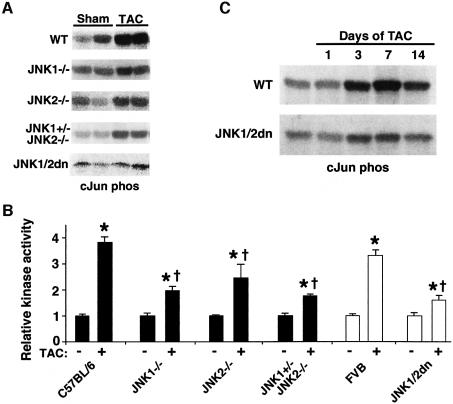
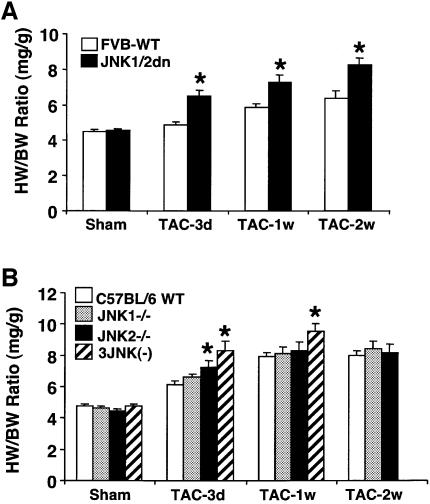
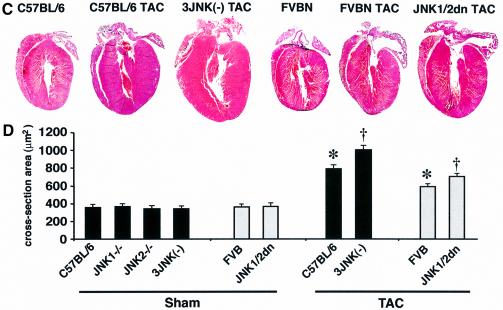
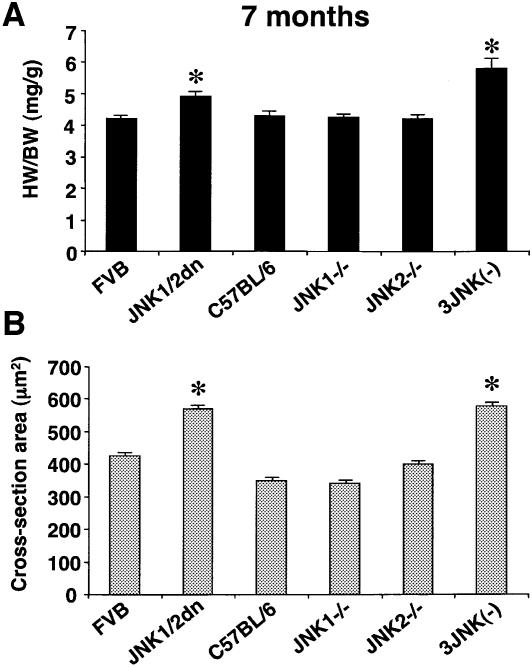
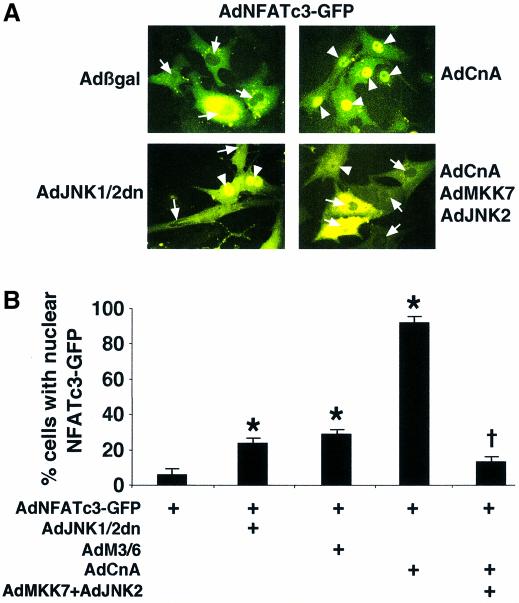
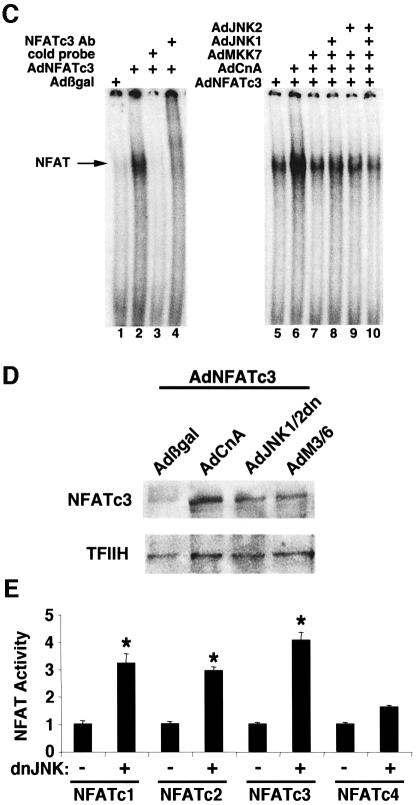
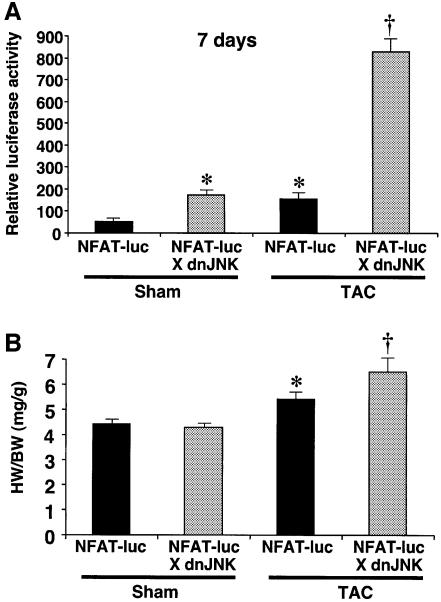
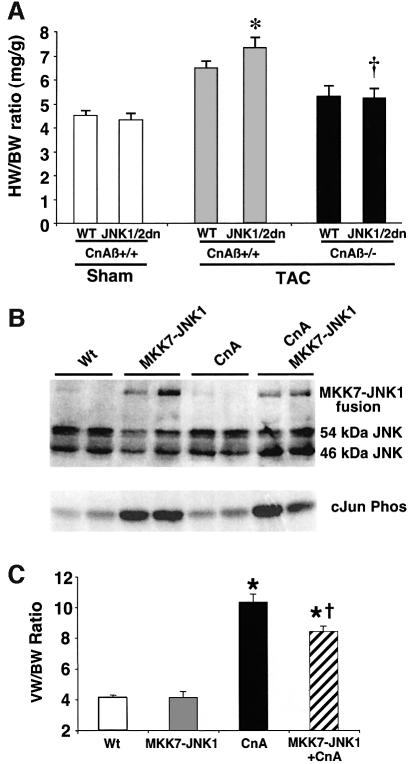
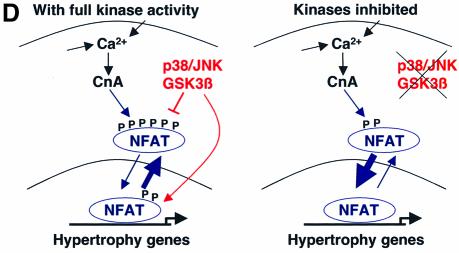
The c-Jun N-terminal kinase (JNK) branch of the mitogen-activated protein kinase (MAPK) signaling pathway regulates cellular differentiation, stress responsiveness and apoptosis in multicellular eukaryotic organisms. Here we investigated the functional importance of JNK signaling in regulating differentiated cellular growth in the post-mitotic myocardium. JNK1/2 gene-targeted mice and transgenic mice expressing dominant negative JNK1/2 were determined to have enhanced myocardial growth following stress stimulation or with normal aging. A mechanism underlying this effect was suggested by the observation that JNK directly regulated nuclear factor of activated T-cell (NFAT) activation in culture and in transgenic mice containing an NFAT-dependent luciferase reporter. Moreover, calcineurin Abeta gene targeting abrogated the pro-growth effects associated with JNK inhibition in the heart, while expression of an MKK7-JNK1 fusion protein in the heart partially reduced calcineurin-mediated cardiac hypertrophy. Collectively, these results indicate that JNK signaling antagonizes the differentiated growth response of the myocardium through direct cross-talk with the calcineurin-NFAT pathway. These results also suggest that myocardial JNK activation is primarily dedicated to modulating calcineurin-NFAT signaling in the regulation of differentiated heart growth.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
