

Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene
- PMID: 14519688
- PMCID: PMC2862298
- DOI: 10.1093/hmg/ddg332
Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene
Abstract
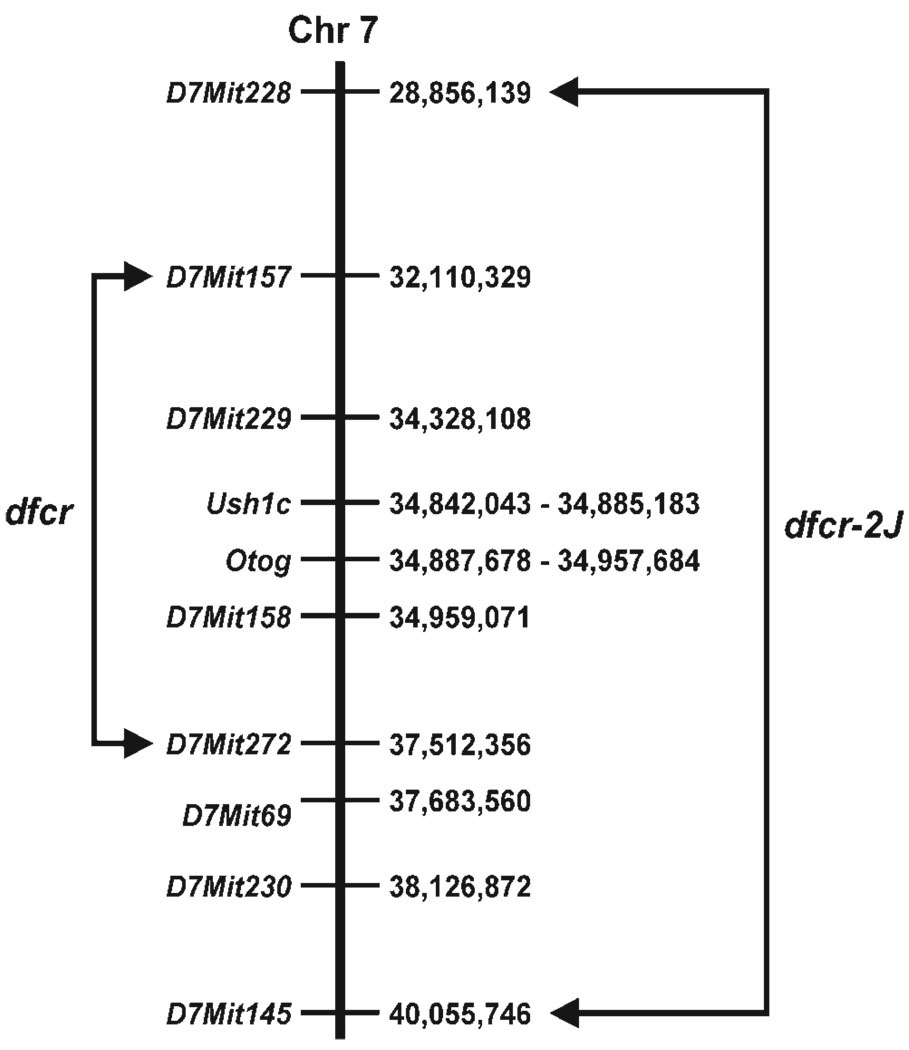
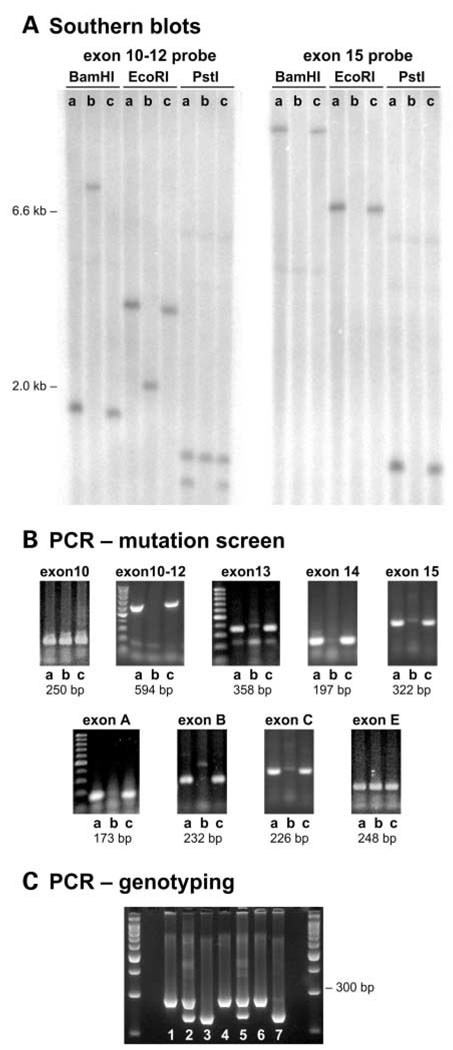
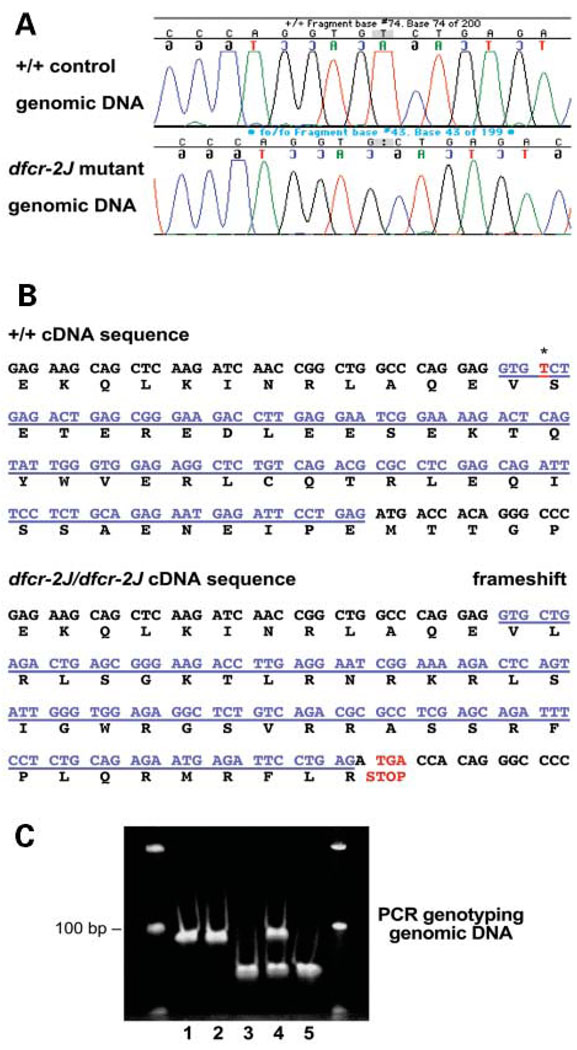
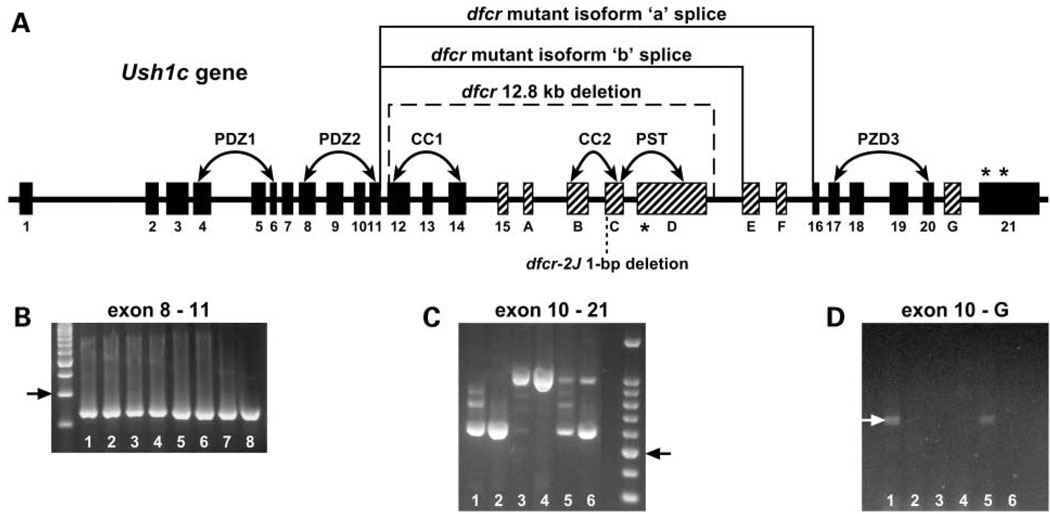
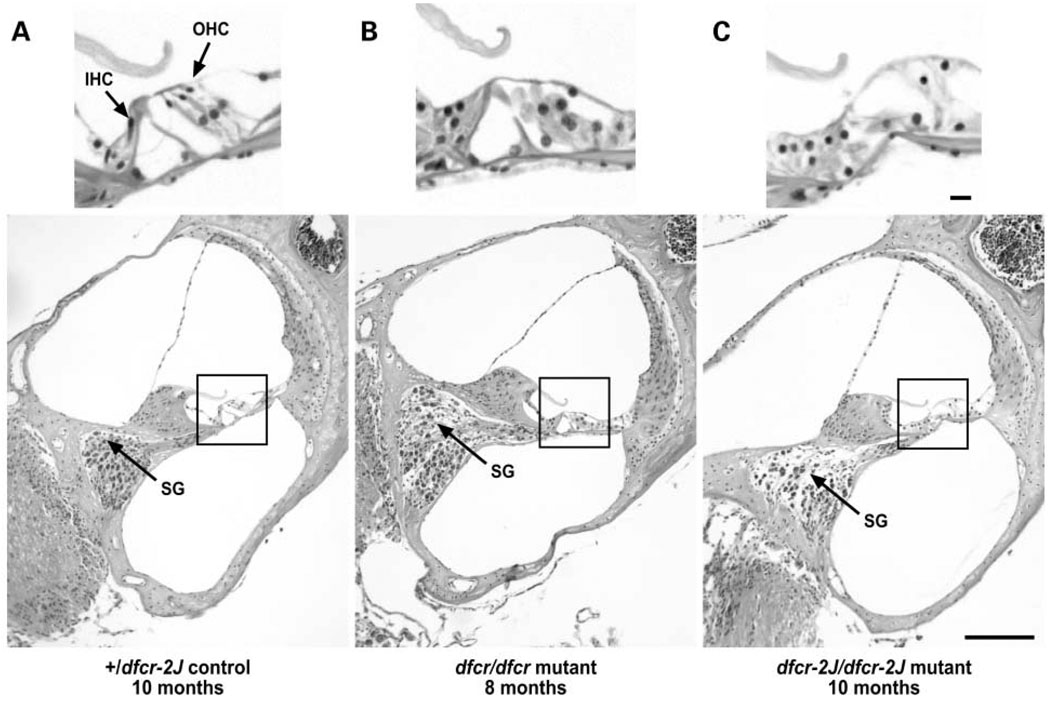
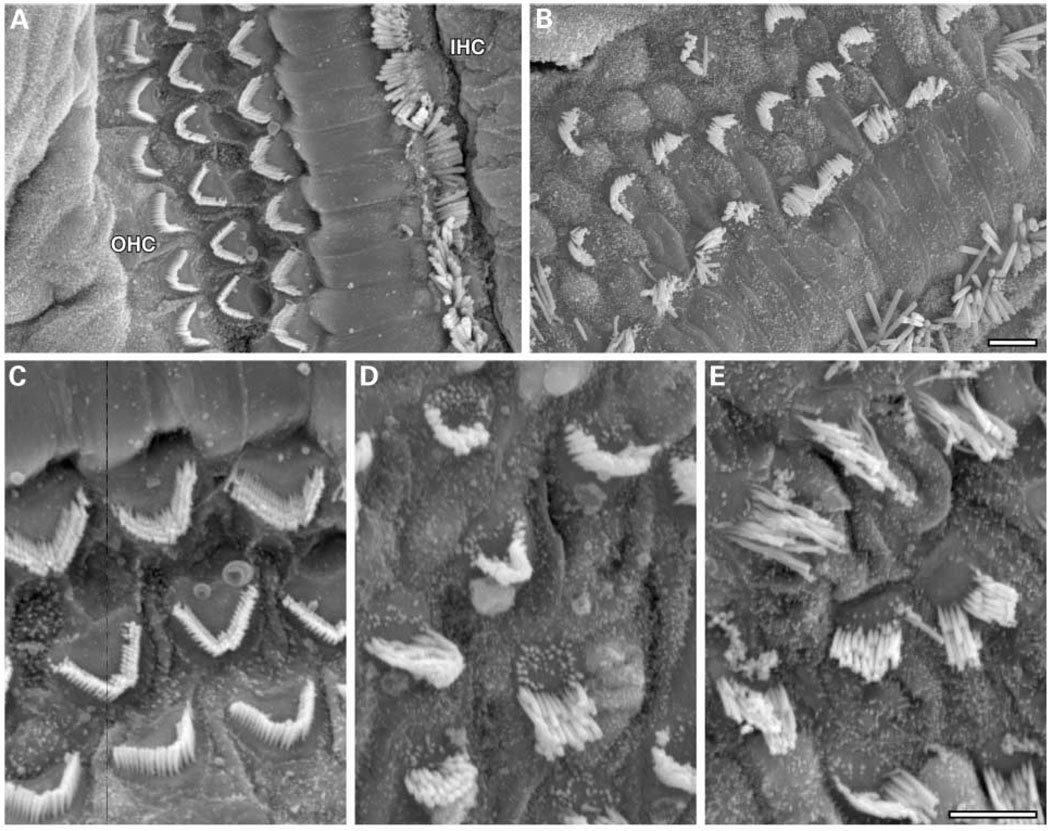
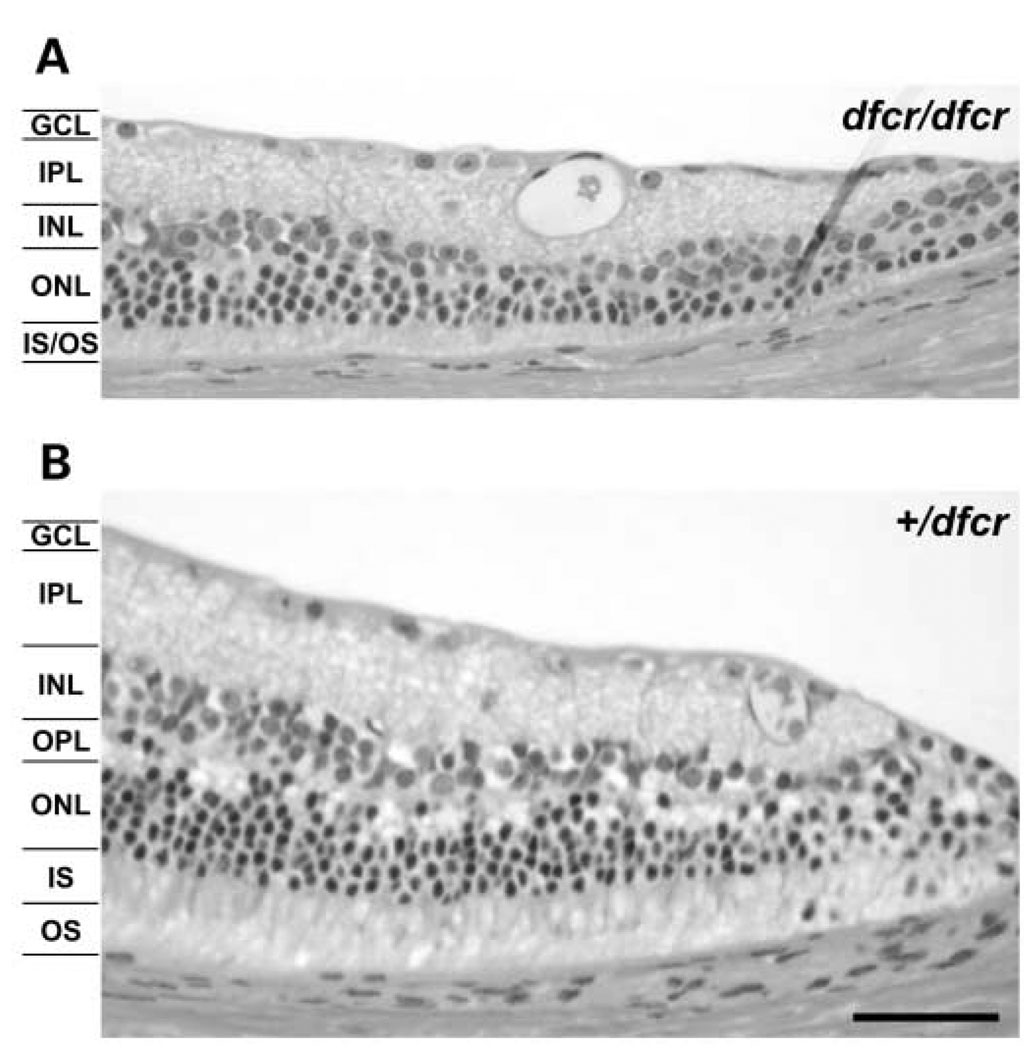
We mapped two new recessive mutations causing circling behavior and deafness to the same region on chromosome 7 and showed they are allelic by complementation analysis. One was named 'deaf circler' (allele symbol dfcr) and the other 'deaf circler 2 Jackson' (allele symbol dfcr-2J). Both were shown to be mutations of the Ush1c gene, the mouse ortholog of the gene responsible for human Usher syndrome type IC and for the non-syndromic deafness disorder DFNB18. The Ush1c gene contains 28 exons, 20 that are constitutive and eight that are alternatively spliced. The dfcr mutation is a 12.8 kb intragenic deletion that eliminates three constitutive and five alternatively spliced exons. The dfcr-2J mutation is a 1 bp deletion in an alternatively spliced exon that creates a transcriptional frame shift, changing 38 amino acid codons before introducing a premature stop codon. Both mutations cause congenital deafness and severe balance deficits due to inner ear dysfunction. The stereocilia of cochlear hair cells are disorganized and splayed in mutant mice, with subsequent degeneration of the hair cells and spiral ganglion cells. Harmonin, the protein encoded by Ush1c, has been shown to bind, by means of its PDZ-domains, with the products of other Usher syndrome genes, including Myo7a, Cdh23 and Sans. The complexes formed by these protein interactions are thought to be essential for maintaining the integrity of hair cell stereocilia. The Ush1c mutant mice described here provide a means to directly investigate these interactions in vivo and to evaluate gene structure-function relationships that affect inner ear and eye phenotypes.
Figures
References
-
- Smith RJ, Lee EC, Kimberling WJ, Daiger SP, Pelias MZ, Keats BJ, Jay M, Bird A, Reardon W, Guest M, et al. Localization of two genes for Usher syndrome type I to chromosome 11. Genomics. 1992;14:995–1002. - PubMed
-
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, et al. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat. Genet. 2000;26:56–60. - PubMed
-
- Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, et al. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet. 2000;26:51–55. - PubMed
-
- Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, Menon PS, Deshmukh D, Griffith AJ, Friedman TB, et al. Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum. Genet. 2002;110:527–531. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RR01183/RR/NCRR NIH HHS/United States
- T32 DK007449/DK/NIDDK NIH HHS/United States
- EY07758/EY/NEI NIH HHS/United States
- P30 CA034196/CA/NCI NIH HHS/United States
- DC04376/DC/NIDCD NIH HHS/United States
- DC62108/DC/NIDCD NIH HHS/United States
- DC04301/DC/NIDCD NIH HHS/United States
- R01 EY007758/EY/NEI NIH HHS/United States
- CA34196/CA/NCI NIH HHS/United States
- R21 DC005846/DC/NIDCD NIH HHS/United States
- HL64885/HL/NHLBI NIH HHS/United States
- DK07449/DK/NIDDK NIH HHS/United States
- R01 DC004301/DC/NIDCD NIH HHS/United States
- R03 DC004376/DC/NIDCD NIH HHS/United States
- R01 HL064885/HL/NHLBI NIH HHS/United States
- P40 RR001183/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
