

Conjugated linoleic acid in humans: regulation of adiposity and insulin sensitivity
- PMID: 14519781
- PMCID: PMC1307498
- DOI: 10.1093/jn/133.10.3041
Conjugated linoleic acid in humans: regulation of adiposity and insulin sensitivity
Abstract
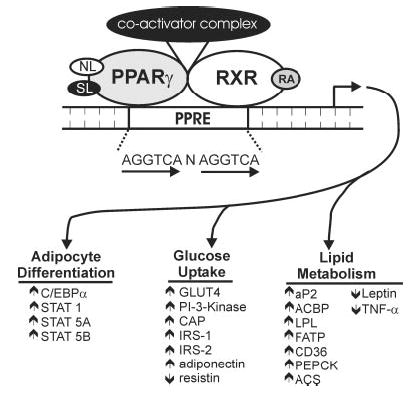
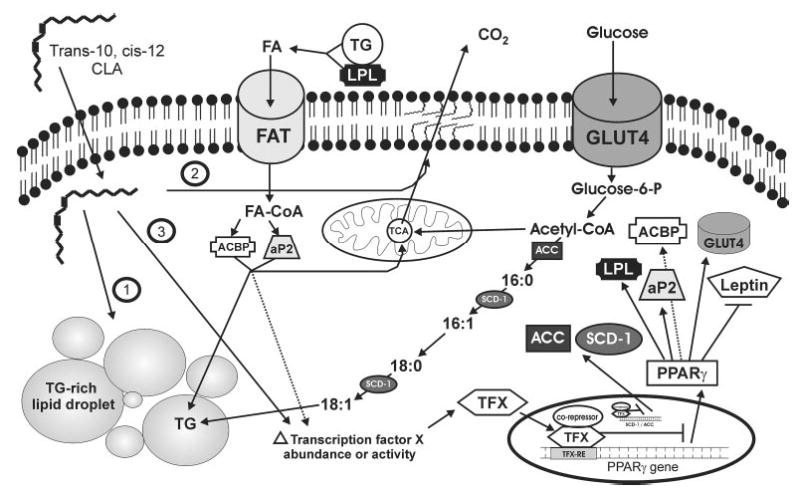
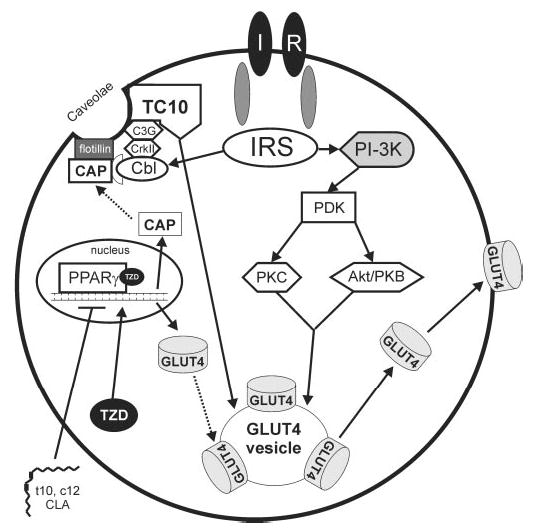
Conjugated linoleic acid (CLA) isomers, a group of positional and geometric isomers of linoleic acid [18:2(n-6)], have been studied extensively due to their ability to modulate cancer, atherosclerosis, obesity, immune function and diabetes in a variety of experimental models. The purpose of this review was to examine CLA's isomer-specific regulation of adiposity and insulin sensitivity in humans and in cultures of human adipocytes. It has been clearly demonstrated that specific CLA isomers or a crude mixture of CLA isomers prevent the development of obesity in certain rodent and pig models. This has been attributed mainly to trans-10, cis-12 CLA, both in vivo and in vitro. However, CLA's ability to modulate human obesity remains controversial because data from clinical trials using mixed isomers are conflicting. In support of some studies in humans, our group demonstrated that trans-10, cis-12 CLA prevents triglyceride (TG) accumulation in primary cultures of differentiating human preadipocytes. In contrast, cis-9, trans-11 CLA increases TG content. Closer examination has revealed that CLA's antiadipogenic actions are due, at least in part, to regulation of glucose and fatty acid uptake and metabolism. This review presents our current understanding of potential isomer-specific mechanisms by which CLA reduces human adiposity and insulin sensitivity.
Figures
References
-
- Ha YL, Grimm NK, Pariza M. Anticarcinogens from fried ground beef: heat-altered derivatives of linoleic acid. Carcinogenesis. 1987;8:1881–1887. - PubMed
-
- Belury M. Inhibition of carcinogenesis by conjugated linoleic acid: potential mechanisms of action. J Nutr. 2002;132:2995–2998. - PubMed
-
- Bassaganya-Riera J, Hontecillas &, Beitz DC. Colonic anti-inflammatory mechanisms of conjugated linoleic acid. Clin Nutr. 2002;2:451–459. - PubMed
-
- Kritchevsky D, Tepper SA, Wright S, Tso P, Czarnecki SK. Influence of conjugated linoleic acid (CLA) on establishment and progression of atherosclerosis in rabbits. J Am Coll Nutr. 2000;19:472S–477S. - PubMed
-
- Belury MA. Dietary conjugated linoleic acid in health: physiological effects and mechanisms of action. Annu Rev Nutr. 2002;22:505–531. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous