

Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A)
- PMID: 14523039
- PMCID: PMC198523
- DOI: 10.1172/JCI18062
Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A)
Abstract
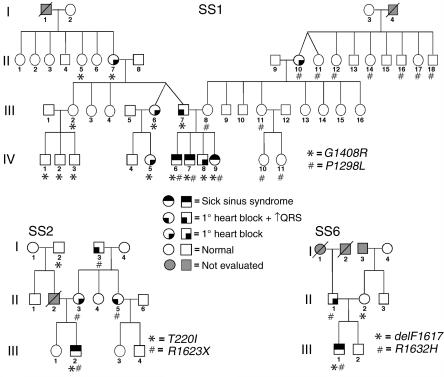
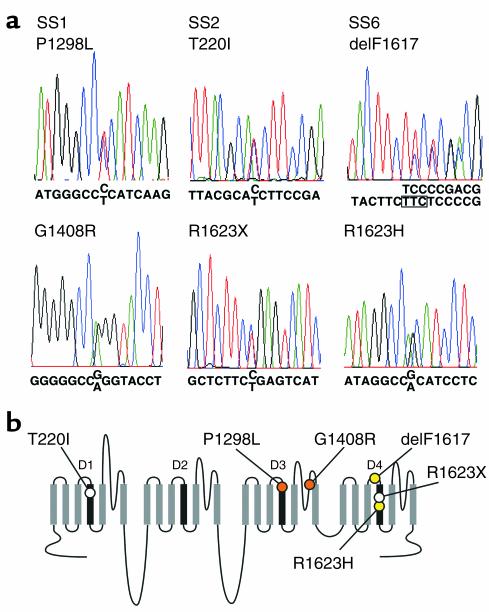
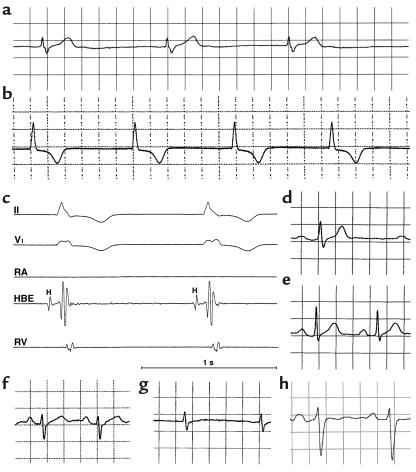
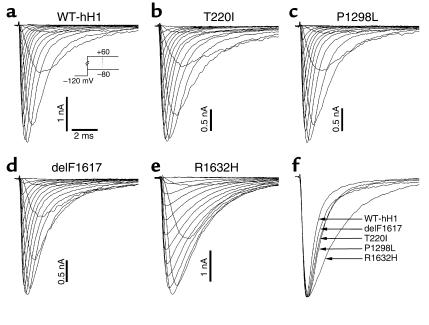
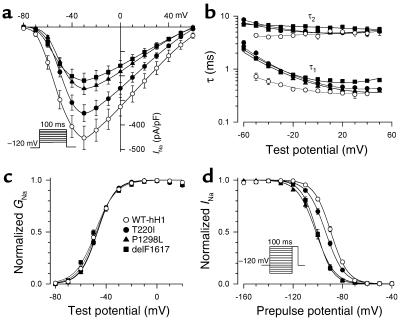
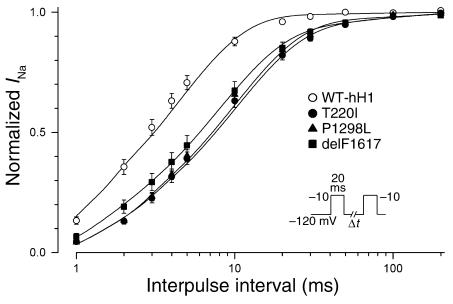
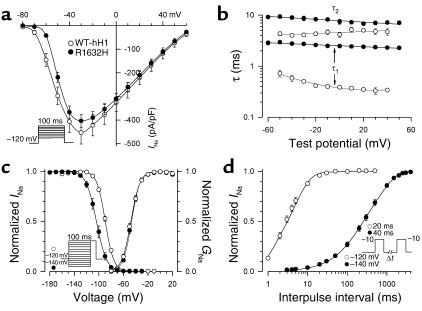
Sick sinus syndrome (SSS) describes an arrhythmia phenotype attributed to sinus node dysfunction and diagnosed by electrocardiographic demonstration of sinus bradycardia or sinus arrest. Although frequently associated with underlying heart disease and seen most often in the elderly, SSS may occur in the fetus, infant, and child without apparent cause. In this setting, SSS is presumed to be congenital. Based on prior associations with disorders of cardiac rhythm and conduction, we screened the alpha subunit of the cardiac sodium channel (SCN5A) as a candidate gene in ten pediatric patients from seven families who were diagnosed with congenital SSS during the first decade of life. Probands from three kindreds exhibited compound heterozygosity for six distinct SCN5A alleles, including two mutations previously associated with dominant disorders of cardiac excitability. Biophysical characterization of the mutants using heterologously expressed recombinant human heart sodium channels demonstrate loss of function or significant impairments in channel gating (inactivation) that predict reduced myocardial excitability. Our findings reveal a molecular basis for some forms of congenital SSS and define a recessive disorder of a human heart voltage-gated sodium channel.
Figures
References
-
- Fish, F.A., and Benson, D.W. 2001. Disorders of cardiac rhythm and conduction. In Moss and Adam’s heart disease in infants, children and adolescents. H.D. Allen, E.B. Clark, H.P. Gutgesell, and D.J. Driscoll, editors. Lippincott, Williams & Wilkins. Philadelphia, Pennsylvania, USA. 482–533.
-
- Martin, A.B., and Kugler, J.D. 1999. Sinus node dysfunction. In Clinical pediatric arrhythmias: electrophysiology and pacing. P.C. Gillette and A.J. Garson, editors. W.B. Saunders. Philadelphia, Pennsylvania, USA. 51–62.
-
- Yabek SM, Swensson RE, Jarmakani JM. Electrocardiographic recognition of sinus node dysfunction in children and young adults. Circulation. 1977;56:235–239. - PubMed
-
- Yabek SM, Jarmakani JM. Sinus node dysfunction in children, adolescents, and young adults. Pediatrics. 1978;61:593–598. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
