

Phylogenetic diversity, abundance, and axial distribution of bacteria in the intestinal tract of two soil-feeding termites (Cubitermes spp.)
- PMID: 14532056
- PMCID: PMC201194
- DOI: 10.1128/AEM.69.10.6007-6017.2003
Phylogenetic diversity, abundance, and axial distribution of bacteria in the intestinal tract of two soil-feeding termites (Cubitermes spp.)
Abstract
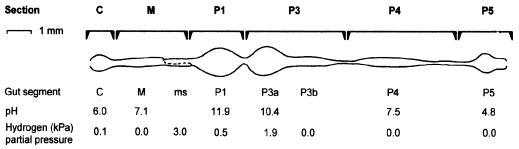
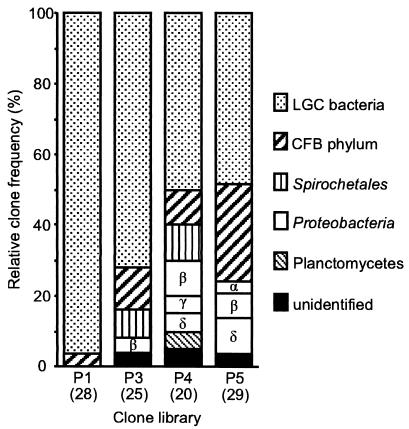
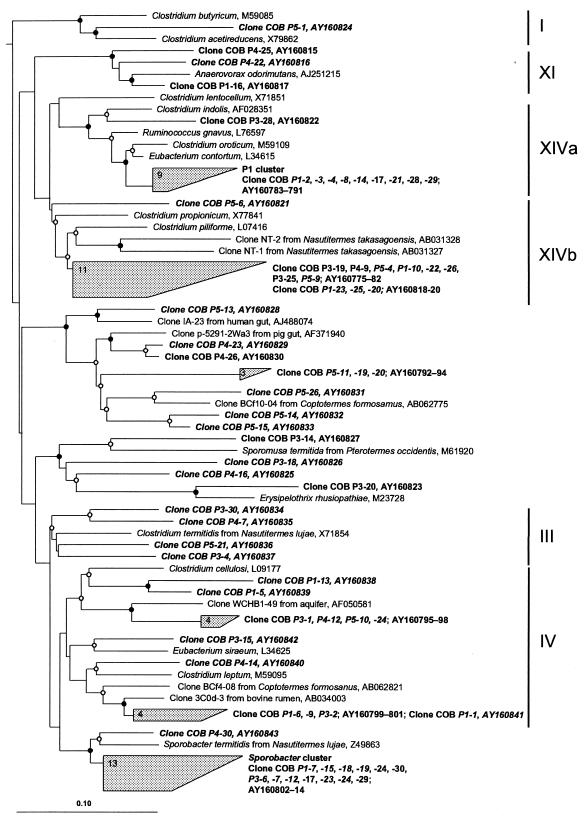
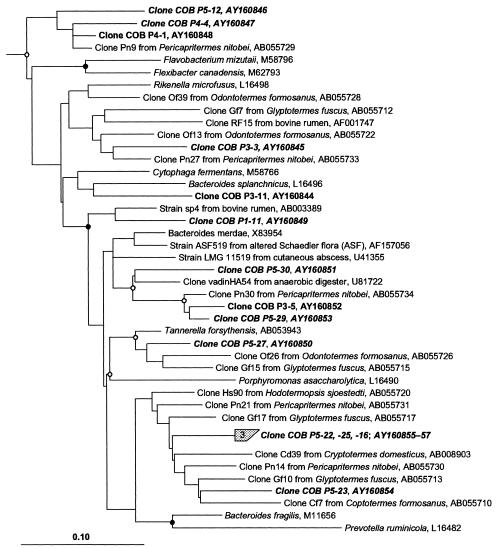
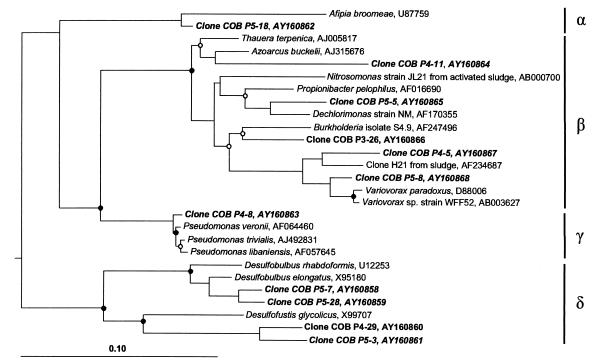
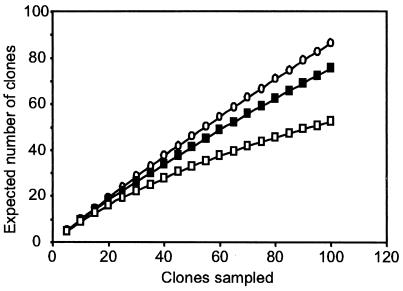
The hindgut of soil-feeding termites is highly compartmentalized and characterized by pronounced axial dynamics of the intestinal pH and microbial processes such as hydrogen production, methanogenesis, and reductive acetogenesis. Nothing is known about the bacterial diversity and the abundance or axial distribution of the major phylogenetic groups in the different gut compartments. In this study, we showed that the variety of physicochemical conditions is reflected in the diversity of the microbial communities in the different gut compartments of two Cubitermes species (TERMITIDAE: Termitinae). 16S rRNA gene clones from the highly alkaline first proctodeal segment (P1) of Cubitermes orthognathus represented almost exclusively gram-positive bacteria with low G+C content (LGC bacteria). In the posterior gut segments, their proportion decreased progressively, and the clone libraries comprised a variety of phyla, including the Cytophaga-Flexibacter-Bacteroides group, various subgroups of Proteobacteria, and the spirochetes. Phylogenetic analysis revealed that many of the clones clustered with sequences from the guts of other termites, and some even formed clusters containing only clones from C. orthognathus. The abundance and axial distribution of major phylogenetic groups in the gut of Cubitermes ugandensis were determined by fluorescence in situ hybridization with group-specific oligonucleotide probes. While the results were generally in good agreement with those of the clonal analysis, direct counts with probes specific for the Planctomycetales revealed a severe underestimation of representatives of this phylum in the clone libraries. Results obtained with newly designed FISH probes directed against two clusters of LGC clones from C. orthognathus indicated that the clones were restricted to specific gut regions. A molecular fingerprinting analysis published in a companion paper (D. Schmitt-Wagner, M. W. Friedrich, B. Wagner, and A. Brune, Appl. Environ. Microbiol. 69:6018-6024, 2003) corroborated the presence of compartment-specific bacterial communities in the gut of different Cubitermes species.
Figures
Similar articles
-
Axial dynamics, stability, and interspecies similarity of bacterial community structure in the highly compartmentalized gut of soil-feeding termites (Cubitermes spp.).Appl Environ Microbiol. 2003 Oct;69(10):6018-24. doi: 10.1128/AEM.69.10.6018-6024.2003. Appl Environ Microbiol. 2003. PMID: 14532057 Free PMC article.
-
Axial differences in community structure of Crenarchaeota and Euryarchaeota in the highly compartmentalized gut of the soil-feeding termite Cubitermes orthognathus.Appl Environ Microbiol. 2001 Oct;67(10):4880-90. doi: 10.1128/AEM.67.10.4880-4890.2001. Appl Environ Microbiol. 2001. PMID: 11571197 Free PMC article.
-
Localization and in situ activities of homoacetogenic bacteria in the highly compartmentalized hindgut of soil-feeding higher termites (Cubitermes spp.).Appl Environ Microbiol. 1999 Oct;65(10):4497-505. doi: 10.1128/AEM.65.10.4497-4505.1999. Appl Environ Microbiol. 1999. PMID: 10508081 Free PMC article.
-
Molecular phylogenetic identification of the intestinal anaerobic microbial community in the hindgut of the termite, Reticulitermes speratus, without cultivation.Extremophiles. 1998 Aug;2(3):155-61. doi: 10.1007/s007920050055. Extremophiles. 1998. PMID: 9783160 Review.
-
[Diversity and function of symbiotic microbes in the gut of lower termites].Wei Sheng Wu Xue Bao. 2006 Jun;46(3):496-9. Wei Sheng Wu Xue Bao. 2006. PMID: 16933630 Review. Chinese.
Cited by
-
Differences between bacterial communities in the gut of a soil-feeding termite (Cubitermes niokoloensis) and its mounds.Appl Environ Microbiol. 2007 Aug;73(16):5199-208. doi: 10.1128/AEM.02616-06. Epub 2007 Jun 15. Appl Environ Microbiol. 2007. PMID: 17574999 Free PMC article.
-
Geographical distribution and diversity of bacteria associated with natural populations of Drosophila melanogaster.Appl Environ Microbiol. 2007 Jun;73(11):3470-9. doi: 10.1128/AEM.02120-06. Epub 2007 Mar 30. Appl Environ Microbiol. 2007. PMID: 17400769 Free PMC article.
-
Soil Macroinvertebrate Presence Alters Microbial Community Composition and Activity in the Rhizosphere.Front Microbiol. 2019 Feb 22;10:256. doi: 10.3389/fmicb.2019.00256. eCollection 2019. Front Microbiol. 2019. PMID: 30853947 Free PMC article.
-
A unique midgut-associated bacterial community hosted by the cave beetle Cansiliella servadeii (Coleoptera: Leptodirini) reveals parallel phylogenetic divergences from universal gut-specific ancestors.BMC Microbiol. 2013 Jun 10;13:129. doi: 10.1186/1471-2180-13-129. BMC Microbiol. 2013. PMID: 23758657 Free PMC article.
-
Development of the human infant intestinal microbiota.PLoS Biol. 2007 Jul;5(7):e177. doi: 10.1371/journal.pbio.0050177. Epub 2007 Jun 26. PLoS Biol. 2007. PMID: 17594176 Free PMC article.
References
-
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. - PubMed
-
- Amann, R., and W. Ludwig. 2000. Ribosomal RNA-targeted nucleic acid probes for studies in microbial ecology. FEMS Microbiol. Rev. 24:555-565. - PubMed
-
- Avgustin, G., R. J. Wallace, and H. J. Flint. 1997. Phenotypic diversity among ruminal isolates of Prevotella ruminicola: proposal of Prevotella brevis sp. nov., Prevotella bryantii sp. nov., and Prevotella albensis sp. nov. and redefinition of Prevotella ruminicola. Int. J. Syst. Bacteriol. 47:284-288. - PubMed
-
- Berchtold, M., A. Chatzinotas, W. Schönhuber, A. Brune, R. Amann, D. Hahn, and H. König. 1999. Differential enumeration and in situ localization of microorganisms in the hindgut of the lower termite Mastotermes darwiniensis by hybridization with rRNA-targeted probes. Arch. Microbiol. 172:407-416. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
