

The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity
- PMID: 14557625
- PMCID: PMC229358
- DOI: 10.1128/jvi.77.21.11398-11407.2003
The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity
Abstract
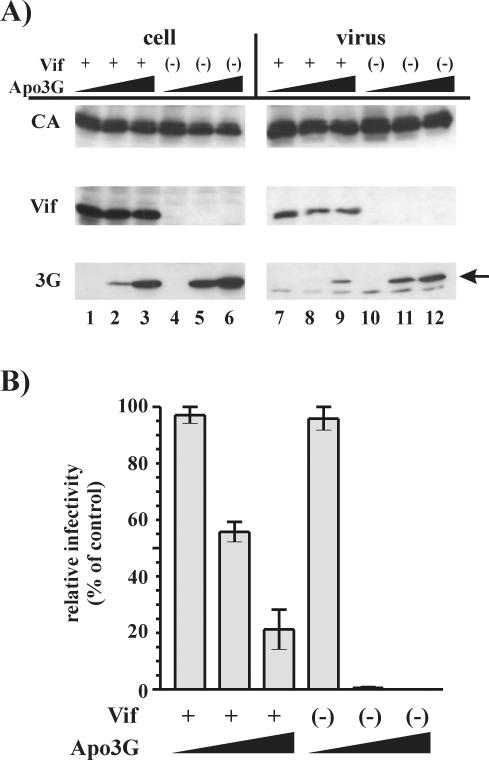
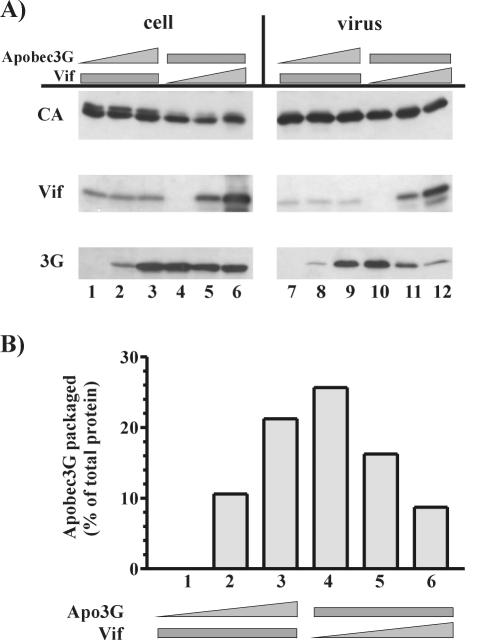
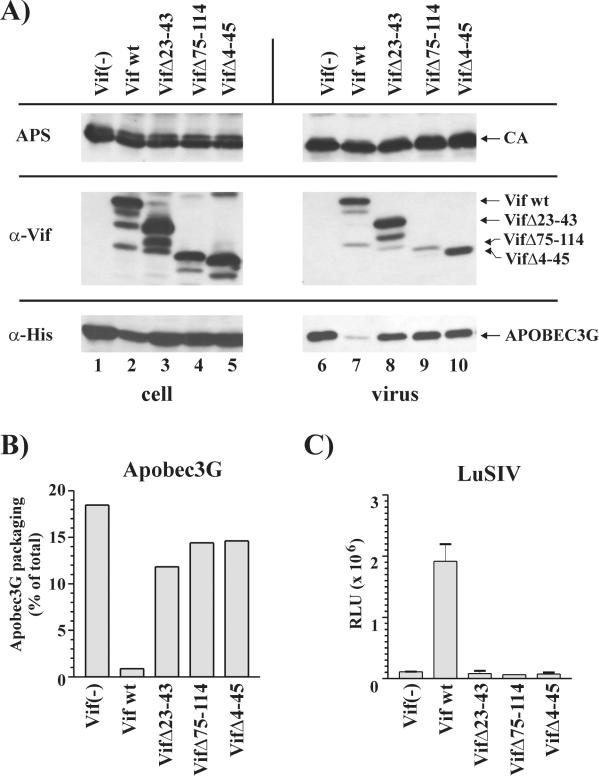
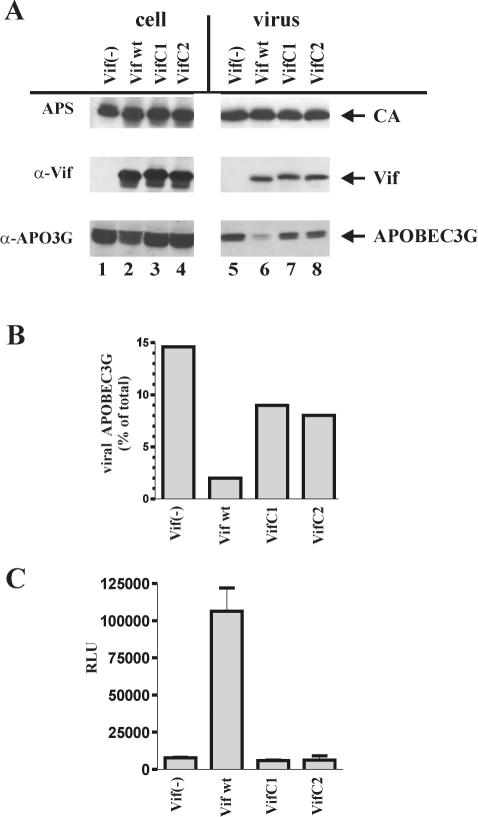
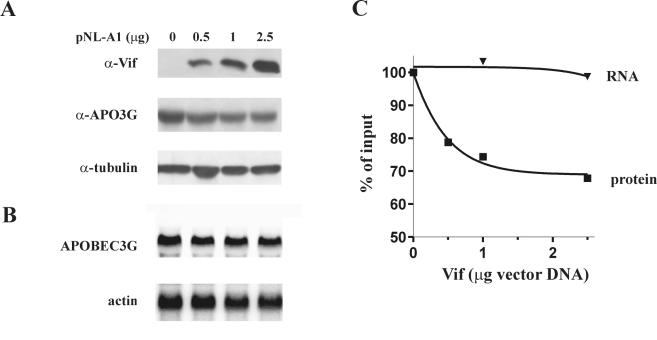
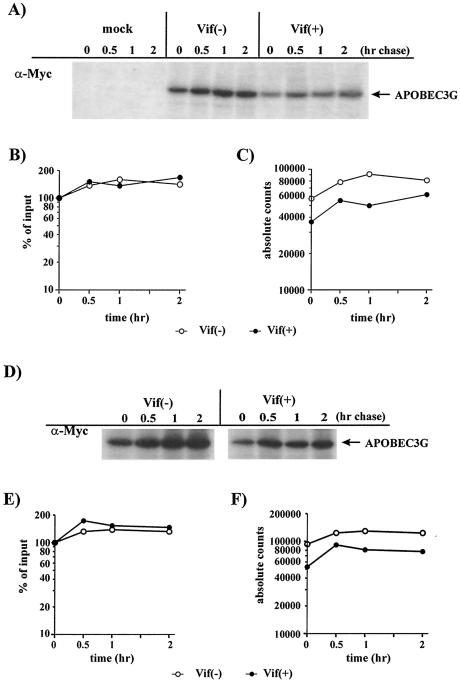
Replication of human immunodeficiency virus type 1 (HIV-1) in most primary cells and some immortalized T-cell lines depends on the activity of the viral infectivity factor (Vif). Vif has the ability to counteract a cellular inhibitor, recently identified as CEM15, that blocks infectivity of Vif-defective HIV-1 variants. CEM15 is identical to APOBEC3G and belongs to a family of proteins involved in RNA and DNA deamination. We cloned APOBEC3G from a human kidney cDNA library and confirmed that the protein acts as a potent inhibitor of HIV replication and is sensitive to the activity of Vif. We found that wild-type Vif inhibits packaging of APOBEC3G into virus particles in a dose-dependent manner. In contrast, biologically inactive variants carrying in-frame deletions in various regions of Vif or mutation of two highly conserved cysteine residues did not inhibit packaging of APOBEC3G. Interestingly, expression of APOBEC3G in the presence of wild-type Vif not only affected viral packaging but also reduced its intracellular expression level. This effect was not seen in the presence of biologically inactive Vif variants. Pulse-chase analyses did not reveal a significant difference in the stability of APOBEC3G in the presence or absence of Vif. However, in the presence of Vif, the rate of synthesis of APOBEC3G was slightly reduced. The reduction of intracellular APOBEC3G in the presence of Vif does not fully account for the Vif-induced reduction of virus-associated APOBEC3G, suggesting that Vif may function at several levels to prevent packaging of APOBEC3G into virus particles.
Figures
References
-
- Akari, H., T. Uchiyama, T. Fukumori, S. Iida, A. H. Koyama, and A. Adachi. 1999. Pseudotyping human immunodeficiency virus type 1 by vesicular stomatitis virus G protein does not reduce the cell-dependent requirement of vif for optimal infectivity: functional difference between Vif and Nef. J. Gen. Virol. 80:2945-2949. - PubMed
-
- Bennett, M. K., N. Calakos, and R. H. Scheller. 1992. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257:255-259. - PubMed
-
- Bour, S., and K. Strebel. 2000. HIV accessory proteins: multifunctional components of a complex system. Adv. Pharmacol. 48:75-120. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
